一测多评法同时测定九味沉香胶囊中9 种成分
2020-06-02李志平王加良张艳丽侯甲福
李志平,王加良∗,张艳丽,张 凯,侯甲福
(1.牡丹江医学院附属红旗医院药学部,黑龙江 牡丹江157011; 2.牡丹江医学院附属红旗医院麻醉科,黑龙江 牡丹江157011; 3.牡丹江医学院药学院,黑龙江 牡丹江157011)
九味沉香胶囊是临床上用于治疗冠心病、心绞痛、脑梗塞属气滞血瘀证的中成药复方制剂,由沉香、广枣、黄芪、川木香、肉豆蔻、当归、西洋参等9 味药材加工而成,方中沉香、川木香、广枣行气止痛、活血安神,合为君药;附以黄芪补气升阳、行滞通痹,当归补血活血,西洋参补气生津,肉豆蔻、诃子温中行气、涩肠止泻,木棉花清热利湿,诸药合用,共奏益气行滞、通络止痛之功效。该制剂现行执行标准为国家药品标准WS-10854(ZD-0854) -2002-2012Z,但仅采用TLC 法对黄芪甲苷进行含有量测定[1],也未检索到相关定量测定的文献报道。
中药及其制剂中有效成分复杂多样,具有多靶点的特点,仅测定单一组分含有量难以对其质量进行全面准确的评价和控制,故多成分或特征性成分定量控制模式已逐步应用于中药及其制剂的质量控制中。一测多评法[2]通过测定制剂中对照品稳定易得的1 种成分,利用中药有效成分间存在的内在函数关系,实现对多成分含有量的同时测定,可有效解决部分对照品不稳定、难以获得等不足,并降低了检验成本,2015 年版《中国药典》 中已有9种中成药应用该方法进行质量控制。因此,本实验采用一测多评法同时测定九味沉香胶囊中沉香四醇、毛蕊异黄酮葡萄糖苷、芒柄花苷、毛蕊异黄酮、芒柄花素、木香羟内酯、去氢木香内酯、肉豆蔻木脂素、去氢二异丁香酚的含有量,以期为全面评价该制剂质量提供依据。
1 材料
1.1 仪器 Agilent 1260 型高效液相色谱仪(美国Agilent 公司),Waters 2695 型高效液相色谱仪(美国Waters 公司);BS210S 型电子天平[赛多利斯科学仪器(北京) 有限公司];KH-300DB 型数控超声仪(昆山禾创超声仪器有限公司)。
1.2 试剂与药物 沉香四醇 (批号111980-201602,纯度98.3%)、毛蕊异黄酮葡萄糖苷(批号111920-201606,纯度97.6%)、去氢二异丁香酚(批号110749-201919,纯度97.1%) 对照品均购自中国食品药品检定研究院;芒柄花苷(批号PRF8081121,纯度99.6%)、毛蕊异黄酮 (批号PRF8062601,纯 度99.6%)、芒柄花 素 (批 号PRF8091225,纯度99.9%) 对照品均购自成都普瑞法科技开发有限公司;肉豆蔻木脂素 (批号CFS201901,纯度98.0%)、木香羟内酯 (批号CFS201802,纯度98.1%)、去氢木香内酯(批号CFS201801,纯度98.0%) 均购自武汉天植生物技术有限公司。沉香、肉豆蔻、广枣、当归、川木香、黄芪、西洋参、诃子、木棉花均购自黑龙江国圣堂中药饮片有限公司,经黑龙江省牡丹江医学院药学院侯甲福副教授鉴定为正品,按均符合相关质量标准检验规定。九味沉香胶囊(每粒装0.3 g,批号1901202、1903218、1905203) 购自青海省格拉丹东药业有限公司。乙腈、甲醇为色谱纯(美国Tedia 公司);其余试剂均为分析纯。
2 方法与结果
2.1 对照品溶液制备 精密称取沉香四醇、毛蕊异黄酮葡萄糖苷、芒柄花苷、毛蕊异黄酮、芒柄花素、木香羟内酯、去氢木香内酯、肉豆蔻木脂素、去氢二异丁香酚对照品适量,甲醇分别制成0.278、0.434、0.252、0.178、0.732、2.854、1.696、0.472、0.138 mg/mL 贮备液,精密吸取适量,甲醇定容,即得(含13.9 μg/mL 沉香四醇、21.7 μg/mL 毛蕊异黄酮葡萄糖苷、12.6 μg/mL 芒柄花苷、8.9 μg/mL 毛蕊异黄酮、36.6 μg/mL 芒柄花素、142.7 μg/mL 木香羟内酯、84.8 μg/mL去氢木香内酯、23.6 μg/mL 肉豆蔻 木脂素、6.9 μg/mL 去氢二异丁香酚)。
2.2 供试品溶液制备 取九味沉香胶囊适量,倒出内容物,研细,精密称取1.0 g,精密加入25 mL甲醇,称 定质量,超 声 (功 率300 W,频 率40 kHz) 处理30 min,放冷,甲醇补足减失的质量,摇匀,滤过,取续滤液,经0.45 μm 微孔滤膜过滤,即得。
2.3 阴性样品溶液制备 按处方比例及生产工艺,分别制备缺沉香、缺黄芪、缺川木香、缺肉豆蔻的阴性样品,按“2.2” 项下方法制备,即得。
2.4 色谱条件与专属性考察 Agilent Zorbax SB-C18色谱柱(250 mm×4.6 mm,5 μm);流动相[乙腈-甲醇(9 ∶1)] (A) -0.1%甲酸(B),梯度洗脱(0~11.0 min,17.0%A;11.0~17.0 min,17.0%~26.0% A;17.0~35.0 min,26.0%~52.0% A;35.0~51.0 min,52.0%~66.0% A;51.0~64.0 min,66.0%~ 72.0% A;64.0~75.0 min,72.0%~ 17.0% A );体积流 量1.0 mL/min;0~35.0 min 在254 nm 波长处检测沉香四醇、毛蕊异黄酮葡萄糖苷、芒柄花苷、毛蕊异黄酮、芒柄花素[3-9],35.0~51.0 min 在225 nm 波长处检测 木香羟内酯、去氢木 香内酯[5,10-12];51.0~75.0 min 在270 nm 波长处检测肉豆蔻木脂素、去氢二异丁香酚[5,13-14];柱温30 ℃;进样量10 μL。取对照品、供试品、阴性样品溶液,在“2.4” 项色谱条件下进样测定,结果见图1,可知各成分色谱峰与相邻峰均能达到有效分离(分离度均>1.5),理论塔板数按各成分计均不低于5 000,阴性无干扰。

图1 各成分HPLC 色谱图Fig.1 HPLC chromatograms of various constituents
2.5 线性关系考察 精密吸取“2.1” 项下贮备液各适量,甲醇制成20 倍质量浓度差的6 个混合标准溶液A~F,在“2.4” 项色谱条件下进样测定。以溶液质量浓度为横坐标(X),峰面积为纵坐标(Y) 进行回归,结果见表1,可知各成分在各自范围内线性关系良好。

表1 各成分线性关系Tab.1 Linear relationships of various constituents
2.6 精密度试验 精密吸取同一供试品溶液,在“2.4” 项色谱条件下进样测定6 次,测得沉香四醇、毛蕊异黄酮葡萄糖苷、芒柄花苷、毛蕊异黄酮、芒柄花素、木香羟内酯、去氢木香内酯、肉豆蔻木脂素、去氢二异丁香酚峰面积RSD 分别为0.92%、0.75%、1.21%、1.18%、0.64%、0.56%、0.53%、0.87%、1.33%,表明仪器精密度良好。
2.7 重复性试验 取同一批胶囊,按“2.2” 项下方法平行制备6 份供试品溶液,在“2.4” 项色谱条件下进样测定,测得沉香四醇、毛蕊异黄酮葡萄糖苷、芒柄花苷、毛蕊异黄酮、芒柄花素、木香羟内酯、去氢木香内酯、肉豆蔻木脂素、去氢二异丁香酚含有量 RSD 分别为 1.31%、1.46%、0.79%、1.63%、1.24%、1.16%、1.07%、1.55%、0.83%,表明该方法重复性良好。
2.8 稳定性试验 取胶囊适量,按“2.2” 项下方法制备供试品溶液,于0、2、4、6、12、18 h在“2.4” 项色谱条件下进样测定,测得沉香四醇、毛蕊异黄酮葡萄糖苷、芒柄花苷、毛蕊异黄酮、芒柄花素、木香羟内酯、去氢木香内酯、肉豆蔻木脂素、去氢二异丁香酚色谱峰峰面积RSD 分别为0.86%、0.79%、1.15%、1.20%、0.68%、0.53%、0.57%、0.85%、1.31%,表明供试品溶液在18 h内稳定性良好。
2.9 加样回收率试验 取含有量已知的胶囊适量,倾出内容物,研细,精密称取9 份,每份0.5 g,分别精密加入对照品溶液(含0.186 mg/mL 沉香四醇、0.248 mg/mL 毛蕊异 黄酮葡 萄糖苷、0.142 mg/mL芒柄花苷、0.108 mg/mL 毛蕊异黄酮、0.446 mg/mL 芒柄花素、1.972 mg/mL 木香羟内酯、1.154 mg/mL 去氢木香内酯、0.272 mg/mL 肉豆蔻木脂素、0.098 mg/mL 去氢二异丁香酚) 0.5、1.0、1.5 mL,各3 份,按“2.2” 项下方法制备供试品溶液,在“2.4” 项色谱条件下进样测定,计算回收率。结果,沉香四醇、毛蕊异黄酮葡萄糖苷、芒柄花苷、毛蕊异黄酮、芒柄花素、木香羟内酯、去氢木香内酯、肉豆蔻木脂素、去氢二异丁香酚平均加样回收 率分别 为 98.38%、99.26%、96.95%、97.55%、99.77%、100.12%、98.67%、97.99%、98.06%,RSD 分别为 1.12%、0.73%、1.22%、1.51%、0.82%、0.91%、1.33%、1.06%、1.36%。
2.10 相对校正因子计算 取“2.5” 项下混合标准溶液A~F,在“2.4” 项色谱条件下进样测定,以木香羟内酯为内标,计算其他8 种成分的相对校正因子,结果见表2。
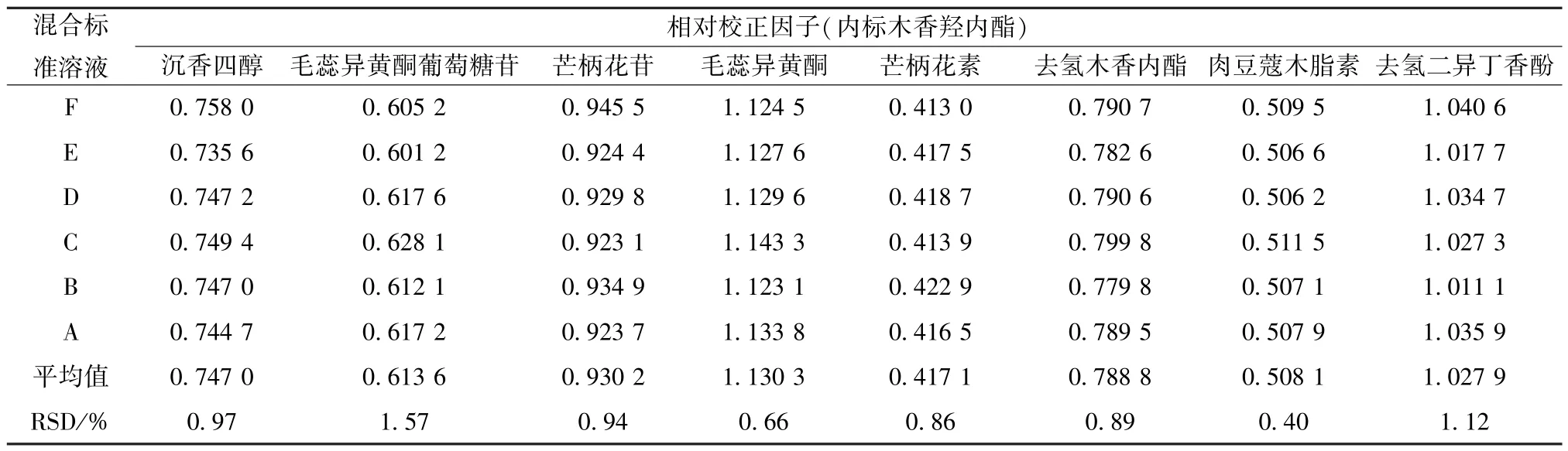
表2 各成分相对校正因子Tab.2 Relative correction factors of various constituents
2.11 耐用性考察
2.11.1 仪器、色谱柱 本实验比较了Agilent 1260 型、Waters 2695 型色谱 仪,以 及Agilent Zorbax SB-C18(250 mm×4.6 mm,5 μm)、Kromasil C18(250 mm×4.6 mm,5 μm)、Waters Symmetry C18(250 mm×4.6 mm,5 μm) 色谱柱对相对校正因子的影响,结果见表3,可知均无明显影响(RSD<2.0%)。
2.11.2 柱温 本实验比较了柱温25、28、30、32、35 ℃) 对相对校正因子的影响,结果见表4,可知均无明显影响(RSD<2.0%)。
2.12 色谱峰定位 以内标(木香羟内酯) 为基准峰,计算“2.11.1” 项下仪器、色谱柱中8 种成分的相对保留时间,结果见表5,可知均无明显影响(RSD<2.0%)。
2.13 样品含有量测定 取3 批胶囊适量,倾出内容物,按“2.2” 项下方法制备供试品溶液,各平行3 份,在“2.4” 项色谱条件下进样测定,计算含有量,结果见表6,可知一测多评法所得结果与外标法接近,相对平均偏差(RAD) 均<2.0%。
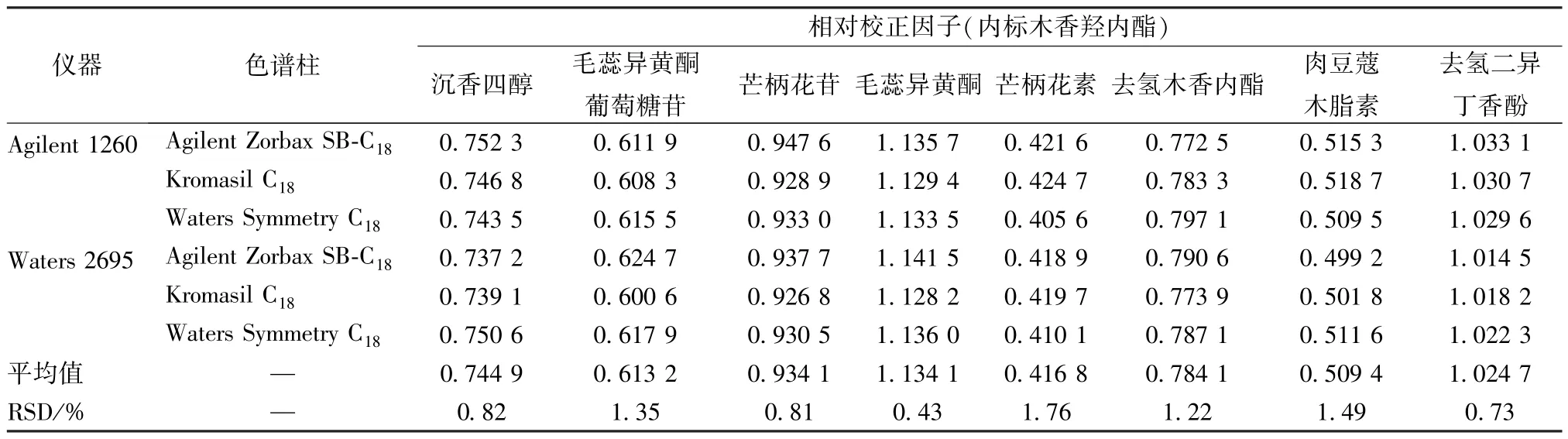
表3 不同仪器、色谱柱对相对校正因子的影响Tab.3 Effects of different instruments and columns on relative correction factors
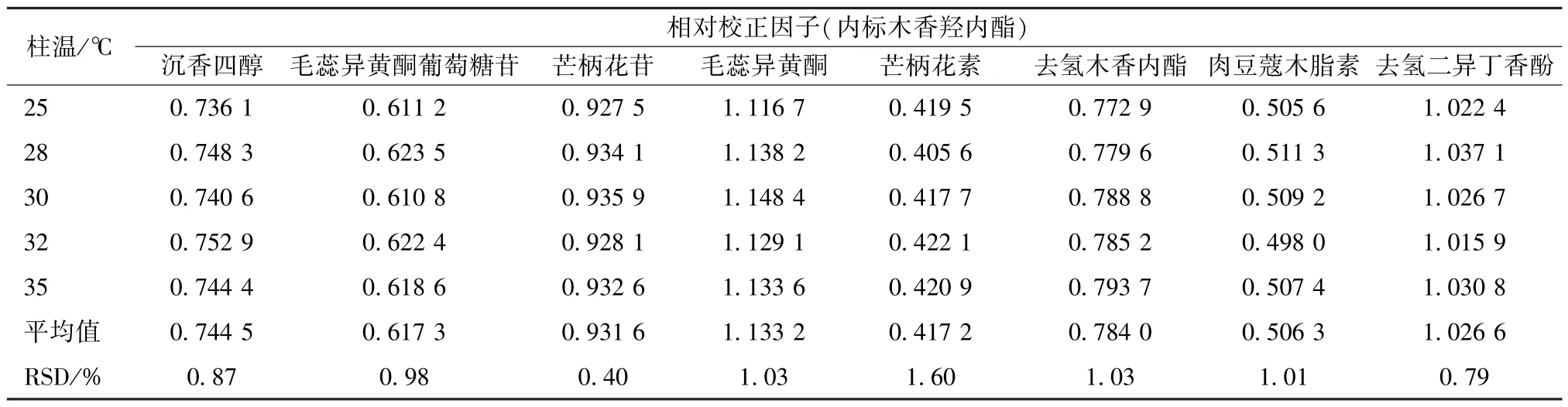
表4 不同柱温对相对校正因子的影响Tab.4 Effects of different column temperatures on relative correction factors

表5 各成分相对保留时间Tab.5 Relative retention time of various constituents

表6 各成分含有量测定结果(mg/粒, n=3)Tab.6 Results of content determination of various constituents (mg/capsule, n=3)
3 讨论
3.1 梯度洗脱条件筛选 本实验首先考察了流动相甲醇-水[5,13]、乙腈-水、[乙腈-甲醇(9 ∶1)] -水[6]对各成分的分离效果,发现以[乙腈-甲醇(9 ∶1)] -水洗脱时较理想,但存在基线不平稳、沉香四醇和毛蕊异黄酮色谱峰分离度不佳等不足,故水相采用一定浓度的酸(0.1%甲酸[3-5,7]、0.1%磷酸[14]、0.1% 冰醋酸),并对有机相、水相比例不断进行摸索,最终确定采用 [乙腈-甲醇(9 ∶1)] 与0.1%甲酸作为流动相,按“2.4” 项下比例进行梯度洗脱,此时各成分均能达到有效分离,而且峰形对称。
3.2 提取方法筛选 本实验在制备供试品溶液时,考察了提取溶剂(75%甲醇[14]、甲醇[5-6,10]、75%乙醇[13]、乙 醇[3-5,7])、提取方 法 (超 声[3-6,10-11]、加热回流[5,7])、提取时间(20、30、40 min),发现甲醇超声提取30 min 时各成分综合提取率最佳,而且杂质峰较少。
4 结论
本实验建立一测多评法对九味沉香胶囊中沉香四醇、毛蕊异黄酮葡萄糖苷、芒柄花苷、毛蕊异黄酮、芒柄花素、木香羟内酯、去氢木香内酯、肉豆蔻木脂素、去氢二异丁香酚含有量同时进行测定,通过耐用性考察、色谱峰定位,并与外标法所得结果进行比较,发现该方法操作简便,结果准确,重复性好,与外标法实测值无明显差异,可为全面评价该制剂质量提供参考。
