多种分子检测方法在精确诊断2例罕见α-珠蛋白生成障碍性贫血中的应用*
2020-05-29姚翠泽王继成秦丹卿袁腾龙黄华洁
姚翠泽,王继成,秦丹卿,杜 丽,刘 玲,袁腾龙,梁 杰,黄华洁
(广东省妇幼保健院医学遗传中心,广东广州 511442)
珠蛋白生成障碍性贫血常见于我国南方地区,是一类因珠蛋白合成障碍所致的遗传性溶血性疾病,主要分为α-珠蛋白生成障碍性贫血和β-珠蛋白生成障碍性贫血两类。α-珠蛋白生成障碍性贫血的分子基础是α2或α1珠蛋白基因发生缺失或点突变致使α-珠蛋白产量下降,其中缺失型约占80%~90%[1-2]。其缺失范围从几kb到整个α-珠蛋白基因簇及上游调控区域的缺失。中国人群中的缺失型α-珠蛋白生成障碍性贫血主要为--SEA、-α3.7、-α4.2三种常见类型[3]。目前,常见的珠蛋白生成障碍性贫血缺失类型出生缺陷防控工作效果显著,但值得注意的是,近年来,我国陆续有多种罕见缺失型α-珠蛋白生成障碍性贫血的研究报道[4-5]。这些罕见缺失型α-珠蛋白生成障碍性贫血如果合并其他常见的α-珠蛋白基因突变可导致中重型α-珠蛋白生成障碍性贫血,所以在珠蛋白生成障碍性贫血高危地区人群中,重视此类罕见珠蛋白生成障碍性贫血的筛查和检测对更全面地预防珠蛋白生成障碍性贫血出生缺陷工作尤为重要。目前国内临床实验室使用跨越断裂点PCR技术的商业化试剂盒只能检测上述中国人3种常见缺失型α-珠蛋白生成障碍性贫血,对于罕见缺失型α-珠蛋白生成障碍性贫血的基因检测还需要多种分子检测方法的应用。本研究通过对2例不同类型的罕见缺失型α-珠蛋白生成障碍性贫血病例的基因检测,为多种分子检测方法在精确诊断罕见α-珠蛋白生成障碍性贫血中的应用提供新的思路,现报道如下。
1 资料与方法
1.1一般资料 2例均就诊于本院产前诊断科。病例1:男,24岁,血液学表现异常,常规珠蛋白生成障碍性贫血基因检测未见异常。其配偶孕21+1周,常规珠蛋白生成障碍性贫血基因检测为--SEA/αα,胎儿超声诊断为胎儿水肿综合征。病例2:女,28岁,孕29+1周,孕期顺利,无输血史。常规珠蛋白生成障碍性贫血基因检测为--SEA纯合突变,与血液学表现不一致。
1.2方法
1.2.1血液学表型分析 乙二胺四乙酸抗凝管采集所有病例外周血2 mL,使用迈瑞2000全自动血液分析仪进行红细胞参数分析,包括血红蛋白(Hb)、平均红细胞容积(MCV)、平均红细胞血红蛋白量(MCH)、红细胞体积分布变异系数(RDW)等,采用快速电泳分析系统Sebia capillary进行各种Hb组分分析,主要包括HbA和HbA2。
1.2.2常规珠蛋白生成障碍性贫血基因检测 采用磁珠法提取患者外周血基因组DNA(试剂盒购自中国厦门致善生物公司);应用跨越断裂点PCR法检测常见的3种α-珠蛋白生成障碍性贫血大片段缺失(-α3.7/、-α4.2/、--SEA/),以及反向斑点杂交法检测3种α-珠蛋白生成障碍性贫血点突变(αQSα/、αCSα/、αWSα/)及17种β-珠蛋白生成障碍性贫血点突变(CD41-42、IVS-Ⅱ-654、-29、-28、CD71-72、CD17、CD43、CD26、CD27-28、CD31、-32、-30、CD14-15、IVS-Ⅰ-1、IVS-Ⅰ-5、Int、Cap),根据电泳条带大小和膜条检测位点出现信号有无来确定缺失类型或点突变类型(试剂盒购自中国深圳亚能生物技术有限公司)。
1.2.3罕见α-珠蛋白生成障碍性贫血基因检测 将提取好的患者DNA使用试剂盒提供的引物进行扩增,扩增产物经过琼脂糖凝胶电泳,检测--THAI、-α27.6、-α21.93种缺失型突变;多重连接依赖性探针扩增(MLPA)用于检测患者珠蛋白生成障碍性贫血α基因簇的拷贝数变化,并应用Coffalyser软件进行数据分析;将患者DNA进行酶切、连接、扩增、纯化、片段化、标记及芯片杂交,使用染色体微阵列(CMA)Affymetrix CytoScan 750K技术分析α基因簇所在的16号染色体短臂区域的DNA拷贝数目的变化;α-珠蛋白基因测序用于检测α-珠蛋白基因上其他的点突变,设计引物分别扩增α1和α2珠蛋白基因,PCR产物送中国上海生工生物有限公司进行双向测序。引物序列如下:HBA1 5′-TGG AGG GTG GAG ACG TCC TG-3′和5′-TCC ATC CCC TCC TCC CGC CCT GCC TTT TC-3′;HBA2 5′-GAT GGG CGG GAG TGG AGT-3′和5′-GGA CAG GGG ATG GTT CAG C-3′,扩增产物大小分别为1 181 bp和1 241 bp。
2 结 果
2.1血液学结果 2例病例均为小细胞低色素性贫血,Hb电泳提示HbA2的比例均不同程度的降低,符合α-珠蛋白生成障碍性贫血的血液学表现,见表1。

表1 2例血液学参数和常规α-珠蛋白生成障碍性贫血基因结果
注:参数参考值MCV 82~100 fL;MCH 27~34 pg;Hb 130~175 g/L(男),115~150 g/L(女);RDW 11.5%~14.5%;HbA2 2.7%~3.5%;HbA 94.50%~97.35%。
2.2常规珠蛋白生成障碍性贫血基因检测结果 病例1常规珠蛋白生成障碍性贫血基因检测未检出异常突变。病例2常规α-珠蛋白生成障碍性贫血基因检测的琼脂糖电泳只见一条--SEA条带,无正常对照条带且未检测到αQSα/、αCSα/、αWSα/这3种突变点及其正常对照点。2例病例均未检测到17种β-珠蛋白生成障碍性贫血点突变。见图1A。
2.3罕见α-珠蛋白生成障碍性贫血基因检测结果 使用--THAI、-α27.6、-α21.9试剂盒检测罕见缺失型α-珠蛋白生成障碍性贫血,病例1结果为阴性;病例2为-α27.6/缺失,再结合常规基因检测显示的--SEA/,其初步确认其基因型应为--SEA/-α27.6。基因的测序显示病例1的HBA1结果正常;而病例2的HBA1基因测序结果正常,但无法扩增出HBA2基因目的片段,提示HBA2缺失或引物区域存在突变。MLPA结果显示病例1在探针POLR3K exon3、HBA-HS40 178、HBA-HS40 382、HBZ region up 364、HBZ region up信号均降低约50%,即拷贝数为1,提示区域从HBZ上游3.5 kb至α-珠蛋白基因簇上游的POLR3K基因至少有102.3 kb的杂合缺失。而病例2的MLPA结果显示为HBM区域上游至HBA1 up337拷贝数为零,即纯合缺失,而HBZ上游至HBZP1下游及HBA1up 226至HBQ1基因处拷贝数为1,即杂合缺失。由于(--SEA/)的缺失区域为HBM区域上游至HBQ1基因处,因此推测另一染色体缺失区域为26.4~32.4 kb。见图1B。

注:图A表示病例2常规缺失型α-珠蛋白生成障碍性贫血电泳图;图B表示罕见缺失型α-珠蛋白生成障碍性贫血电泳图;αα/αα表示正常对照;M表示DNA标准带。
图1 跨越断裂点PCR法电泳图
因探针数量的限制,MLPA不能确定病例1的缺失范围,故进行CMA的检测。结果显示病例1为16p13.3(85880-197064)×1,即长度约为111 kb的杂合缺失,缺失区域包含的基因包括POLR3K、SNRNP25、RHBDF1、MPG、NPRL3,与MLPA结果大体一致,同样包含了α-珠蛋白基因簇的调控区域HS40等。见图2。
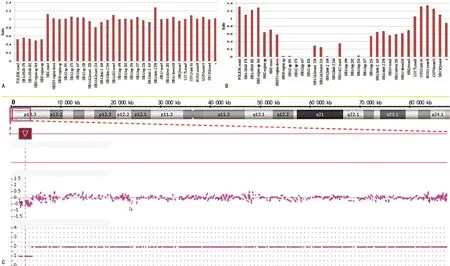
注:A表示病例1MLPA结果分析图;B表示病例2MLPA结果分析图;C表示病例1CMA结果分析图。
图2 两病例MLPA结果和病例1CMA结果分析图
3 讨 论
本研究2病例珠蛋白生成障碍性贫血筛查Hb电泳结果均提示为α-珠蛋白生成障碍性贫血,病例1常规珠蛋白生成障碍性贫血基因检测未检出异常,病例2则检测到基因型与血液学表型不符,均怀疑存在其他罕见缺失,因此在常规基因检测的基础上采用多种分子检测方法进一步精确诊断。应用实验室已有的罕见缺失型α-珠蛋白生成障碍性贫血试剂盒(检测--THAI、-α27.6、-α21.9)和基因测序的方法检测病例1,检测结果均未发现异常,于是采用MLPA检测整个α-珠蛋白基因簇及其上下游调控序列的拷贝数的变化,结果显示α-珠蛋白基因簇上游的POLR3K基因至ζ基因上游区域杂合缺失,缺失片段至少为102.3 kb。因MLPA探针数量较少,为了明确具体缺失范围,同时结合CMA技术进一步验证,结果与MLPA相符,同样为包含HS-40的POLR3K基因至ζ基因的上游区域缺失。目前已有研究证明,调控区域HS-40的缺失会使α-珠蛋白基因表达水平低于正常表达水平的3%,导致α0-珠蛋白生成障碍性贫血,与东南亚型缺失(--SEA/)的血液学表型相同[6-9]。病例2常规跨越断裂点PCR法显示仅有1条SEA条带而没有正常条带(α2),同时基因测序结果显示HBA1测序无异常,而HBA2片段无法扩增获得,考虑到常规珠蛋白生成障碍性贫血显示患者携有(--SEA/)缺失采用MLPA确定缺失的大致范围时,MLPA结果在计算(--SEA/)缺失的信号后推测出另一染色体缺失大小为26.4~32.4 kb,该结果与后续使用罕见缺失型α-珠蛋白生成障碍性贫血试剂盒验证检出的(-27.6/)基因型一致,结合常规和罕见α-珠蛋白生成障碍性贫血的检测结果,可诊断病例2的基因型为(--SEA/-α27.6)。罕见缺失(-α27.6/)的主要缺失范围包括ζ-Ψζ-Ψα2-Ψα1-α2(NG_000006.1:g.9079_36718del27640),仅有1个α1基因发挥功能,为α+-珠蛋白生成障碍性贫血,其杂合子表现正常的Hb水平,MCV、MCH等参数也多接近正常范围的最低值[10-11],其人群携带率仍未有相关报道,但基于其杂合子无临床症状,有理由怀疑这种缺失的携带率比笔者认为的要高。
目前已报道的α-珠蛋白基因簇(NG_000006.1)缺失大约有50种(http://globin.bx.psu.edu/),在中国南方,携带率最高为(--SEA/),占人群4.1%~8.7%[12-13]。本研究中的2例罕见缺失为包含HS-40的POLR3K基因至ζ基因的上游区域缺失和(-α27.6/)缺失,这些罕见型珠蛋白生成障碍性贫血如果使用珠蛋白生成障碍性贫血基因的常规方法检测必然导致其漏诊,且HS-40区域缺失若合并中国南方携带率最高的(--SEA/)缺失时,其后代有25%的概率成为Hb Bart′s水肿胎,同时也可发生孕妇早产、羊水过多、子痫等严重并发症[14]。而罕见缺失(-α27.6/)如果合并(--SEA/)则可导致中间型α-珠蛋白生成障碍性贫血患儿的出生,同时笔者也注意到这种基因型的Hb电泳未见H带,容易造成漏检。对此LING等[15]推测是过剩的β-珠蛋白链发生了不同程度的降解导致。因此,采用多种方法精确诊断罕见α-珠蛋白生成障碍性贫血非常重要。
由于临床上对中重型珠蛋白生成障碍性贫血的治疗主要依靠终身输血和除铁治疗及造血干细胞的移植,其治疗费用昂贵,家庭经济负担沉重,对儿童生长发育影响巨大,珠蛋白生成障碍性贫血患儿出生缺陷“重在预防”。因此,在临床产前遗传咨询中,对于常规珠蛋白生成障碍性贫血基因检测的基因型与血液学表型结果不相符的患者,强烈建议进一步采用多种方法进行精确诊断突变类型,避免夫妇罕见珠蛋白生成障碍性贫血引起中重型珠蛋白生成障碍性贫血患儿的出生。
罕见珠蛋白生成障碍性贫血基因型的精确诊断有助于更加全面、有效地预防中重型珠蛋白生成障碍性贫血患儿的出生,对遗传咨询和产前诊断具有重要指导作用。本研究将为罕见α-珠蛋白生成障碍性贫血的精确诊断提供新思路,为预防出生缺陷工作提供有效的临床诊断基础。
