负载Au 纳米粒子的核壳结构磁性复合催化剂的制备及性能研究
2020-05-22赵晓青
赵晓青
山西工程职业学院,山西 太原 030009
贵金属基纳米催化剂由于其尺寸小、比表面积高而表现出较高的催化性能。贵金属基核壳结构磁性纳米复合材料由磁性核和功能性外壳组成,磁核作为功能性模板决定了最后核壳型微球的尺寸和形态,壳层能将核与外界环境相互隔离,使得核壳型微球的磁核不被外界环境所损坏,因此核壳型微球获得了更广泛的应用。目前,贵金属基磁性核壳结构纳米材料研究主要集中于无机磁性纳米复合材料。DENG 等[1]制备了Fe3O4/n-SiO2/Au/m-SiO2催化材料,其突出优势是磁性复合载体固载Au 纳米粒子后,又包覆一层疏松的介孔二氧化硅层,既保证了活性中心Au 与催化底物的接触,又避免了反应过程中Au 的流失,循环催化8 次后底物转化率依然达到100%,但包覆层的增加使复合催化剂的制备过程变得复杂,也减弱了材料的磁响应性能,降低了回收效率。
贵金属基有机磁性纳米复合材料由磁性有机聚合物材料和贵金属纳米粒子组成,功能性外壳中的有机聚合物随着交联度的增大,耐溶剂性增强,而且表面易于修饰其他功能基团,对贵金属纳米粒子锚固能力增强,因而在催化领域受到了广泛关注。CHANG 等[2]利用天然高分子壳聚糖,制备了Fe3O4@壳聚糖@Au 聚合物磁性纳米复合催化材料,应用于4-硝基苯酚(4-NP)的还原,催化效果良好,可进行磁性回收。XUAN 等[3]对磁核Fe3O4进行表面羧酸化修饰,将苯胺单体与磁核表面的羧基反应交联,通过原位表面聚合法获得了由聚苯胺包覆Fe3O4的磁性载体Fe3O4@PANI(PANI 为聚苯胺),后续还通过静电吸附作用制备了固载Au 的纳米核壳型催化剂Fe3O4@PANI@Au[4],应用于罗丹明B 的降解,表现出较好的催化性能,且利于磁性回收。
有机聚合物材料中的氨基酚醛树脂(APF),由于是体型高分子,在有机溶剂中不容易溶解塌陷,从而可增强复合催化剂的稳定性,另外,APF 表面的-NH2有利于锚固Au 等纳米粒子[5],-OH 有利于其在多种溶剂中分散,提高了Au 等贵金属纳米粒子的利用效率,增加了催化剂的重复利用性。
本工作以聚丙烯酸(PAA,重均分子量为1 800)、3-氨基苯酚、六亚甲基四胺以及Fe3O4磁性粒子为反应物,首先通过水热法对超顺磁性的Fe3O4纳米团簇进行包覆制备得到磁性核壳氨基酚醛树脂微球,然后将其作为催化剂载体在 HAuCl4·4H2O 中经 NaBH4还原固载 Au 纳米粒子获得Fe3O4@APF@Au 磁性纳米复合催化剂,并考察了其在4-硝基苯酚还原反应中的催化性能。此外,还考察了在外加磁场下,复合催化剂的磁分离回收效果和重复使用性能。
1 实验部分
1.1 聚丙烯酸修饰的Fe3O4 纳米团簇的制备
参考李亚栋等[6]提出的溶剂热法制备单分散、单晶的Fe3O4纳米团簇。首先,将2.16 g(8 mmol)FeCl3·6H2O 溶于80 mL 乙二醇中,于60 ℃强磁力搅拌下快速加入0.216 g(0.12 mmol)PAA,磁力搅拌30 min,然后向其中加入18 g 无水乙酸钠,调节转速,强磁力搅拌1 h 后转移到100 mL 反应釜中,在200 ℃条件下反应16 h,最后将其冷却至室温,用去离子水和乙醇交替洗3 次以上,60 ℃条件下真空干燥16 h 待用。
1.2 Fe3O4/氨基酚醛树脂微球(Fe3O4@APF)的制备
采用水热法制备Fe3O4/氨基酚醛树脂微球(Fe3O4@APF)。首先,将溶剂热法制得的聚丙烯酸修饰的Fe3O4纳米团簇0.1 g 超声分散于80 mL 水中,在300 r/min 的机械搅拌下加入一定量的3-氨基苯酚和六亚甲基四胺,室温搅拌30 min 后将反应物转移至100 mL 反应釜中,160 ℃下反应4 h,随后将其冷却至室温,去离子水和乙醇交替洗3 次以上,60 ℃干燥12 h 待用。
1.3 复合催化剂Fe3O4@APF@Au 的制备
称取0.1 g Fe3O4@APF,超声分散于100 mL 水中。冷水浴2 ℃,300 r/min 搅拌下滴入1.8 mL 摩尔浓度为24.3 mmol/L 水合氯金酸(HAuCl4·4H2O),吸附1 h,然后加入4.5 mL 硼氢化钠溶液(0.1 mol/L)还原0.5 h,去离子水和乙醇交替洗3 次,60 ℃干燥12 h,制得Fe3O4@APF@Au 纳米催化剂。
1.4 催化剂表征
采用HITACHI 公司HT7700 型透射电子显微镜(TEM)对溶剂热法制备的Fe3O4@APF@Au 纳米复合催化剂的形貌进行表征;采用日本理学公司SmartLab 型X 射线衍射(XRD)仪分析催化剂的物相和晶型,测试条件:Cu Kα 射线(λ 为1.541 Å),管电压40 kV,管电流200 mA;采用X 射线能谱分析(EDS)表征催化剂中的元素及含量;采用岛津仪器公司DTG-60 型热失重分析仪(TGA)进行热重分析;采用赛默飞世尔科技有限公司Nicolet iS10 型傅里叶变换红外光谱仪(FTIR)对催化剂进行红外光谱测定。
1.5 Fe3O4@APF@Au 纳米复合催化剂催化性能及稳定性测试
采用4-硝基苯酚和硼氢化钠的还原反应来考察复合纳米微球的催化性能。配制摩尔浓度为5 mmol/L的4-硝基苯酚醇溶液,质量分数为1%的催化剂溶液,然后再将催化剂溶液稀释成质量分数分别为0.10%,0.05%和0.01%的溶液。用紫外-可见分光光谱仪(UV-vis,日本岛津公司UV-2550 型)分别测试原始4-硝基苯酚溶液、加入过量硼氢化钠后的4-硝基苯酚溶液以及加入过量硼氢化钠和0.01 mL合适浓度的催化剂溶液,并开始计时,每隔3 min 取样一次并测UV-vis,直至4-硝基苯酚溶液由黄色变为无色为止。
采用紫外分光光度法分析Fe3O4@APF@Au 复合催化剂对4-NP 还原反应的催化活性。通过计算催化底物吸收峰随时间的变化速率得出速率曲线并由此计算催化剂对底物的转化频率(TOF)。转化频率指单位时间单位活性中心上的转化数,其值可以衡量催化剂催化反应的速率,表征催化剂的本征活性。
为测试Fe3O4@APF@Au 纳米复合材料的循环催化性能,取2 mL 摩尔浓度为5 mmol/L 的4-硝基苯酚溶液加入20 mL 水,并加入0.151 8 g 硼氢化钠后摇匀,再加入1 mL 质量分数为1%的催化剂分散液,反应5 min 后进行催化性能测试。磁性分离回收催化剂,经乙醇洗涤数次后用于下次相同条件下的催化反应,如此循环10 次,以测试磁性纳米复合材料的稳定性和循环使用效率。
2 结果与讨论
2.1 Fe3O4/氨基酚醛树脂微球(Fe3O4@APF)的制备及表征
图1(a)是溶剂热法制备的聚丙烯酸修饰的Fe3O4纳米团簇,选用聚丙烯酸修饰是由于聚丙烯酸表面含有大量羧基,可以大大增加Fe3O4微球的亲水性,同时可减弱Fe3O4纳米晶之间的作用力,使其在维持磁响应性的同时保证良好的超顺磁性。由图1(a)可以看到PAA 修饰的Fe3O4纳米团簇呈球形,单分散性好,表面较为粗糙,粒径约为200 nm,排列呈六边形。表面粗糙是由于PAA 修饰的Fe3O4是由数个Fe3O4纳米晶堆砌组成。图1(b)是水热法制备的Fe3O4@APF 复合微球,由TEM 照片可明显看到,以Fe3O4为内核的核壳型结构微球,球形貌光滑,呈单分散分布,APF 树脂成功包覆于Fe3O4纳米团簇表面。

图1 样品的TEM 照片Fig.1 TEM images of samples
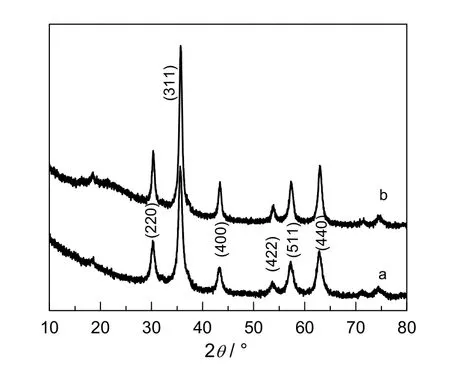
图2 样品的XRD 图谱Fig.2 XRD patterns of samples
图2 是PAA 修饰的Fe3O4纳米团簇和Fe3O4@APF 样品的XRD 图谱。由图2 曲线a 可以看出,在2θ 为30.2,35.6,43.3,53.8,57.5 和62.8°处有6 个比较突出的峰,与标准的JCPDS 卡片相对照,这6 个强峰分别对应具有反尖晶石结构的Fe3O4的(220),(311),(400),(422),(511)和(440)晶面,其结果分析与JCPDS 卡片中的19-0629(Fe3O4)完全符合,说明成功制备出了PAA修饰的Fe3O4。由图2 还可以看出,Fe3O4@APF 复合微球XRD 结果与PAA 修饰的Fe3O4纳米团簇2a比较无明显变化。
PAA 修饰的Fe3O4纳米团簇的红外图谱如图3a 所示,在630 cm-1处的强吸收峰对应着Fe3O4中Fe-O 键的振动吸收,1 407 和1 557 cm-1处的吸收峰分别对应于Fe3O4表面C-O 键的对称振动吸收峰和不对称振动吸收峰,3 430 cm-1处的吸收峰为Fe3O4表面的-OH 的伸缩振动,这表明Fe3O4表面含羧酸基团,证实了Fe3O4表面PAA 的存在,1 630 cm-1处的吸收峰对应于亚甲基的弯曲振动。图3b 是Fe3O4@APF 的红外吸收光谱图,1 237 cm-1的吸收峰是Ar-O-C 的伸缩振动,证实了苯环的存在。
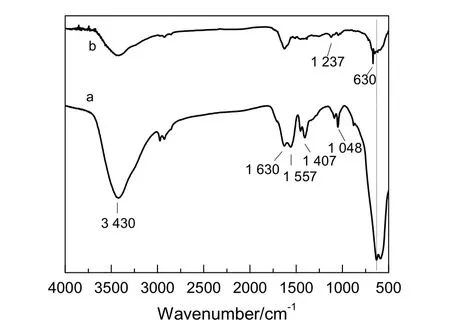
图3 样品的FTIR 图Fig.3 FTIR spectra of samples
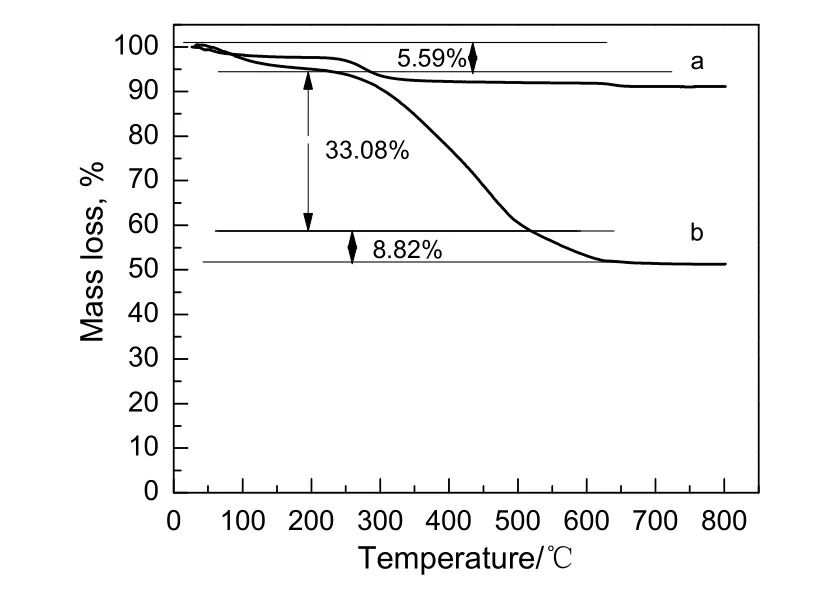
图4 样品的热失重曲线Fig.4 Thermal weight loss curves of samples
对PAA 修饰的Fe3O4纳米团簇和Fe3O4@APF 复合微球进行热重分析,结果如图4 所示。样品由室温加热到800 ℃,PAA 修饰的Fe3O4纳米团簇的重量损失源于纳米颗粒中聚丙烯酸的分解。由图可知,Fe3O4@APF 复合微球的热分解分三个阶段:第一阶段(25~100 ℃)的失重与水和溶剂的蒸发及物理吸附物脱附有关;第二阶段(100~500 ℃)前期的失重是由聚丙烯酸分解所致,当温度从300 ℃升到500 ℃,有30%的重量损失,在这一阶段,APF 酚醛树脂逐渐分解,质量逐渐下降,这源于分解产生了不同挥发性物质(如H2O,CO2,CO,C2H6,H2,CH4和NH3)以及一些乙基衍生物和碳氢化合物;第三阶段(500~627 ℃)有8.82%的重量下降,可归因于Fe3O4纳米颗粒表面APF 链的完全降解。
2.2 复合催化剂Fe3O4@APF@Au 的制备及表征
Fe3O4@APF 复合微球表面-NH2中的氮存在孤对电子,可与Au 纳米粒子上的空轨道形成配位键,因而可作为催化剂载体负载贵金属纳米粒子。Fe3O4@APF@Au 的透射电镜照片如图5 所示。由图可知,负载Au 纳米粒子后,复合微球依然保持球外形和单分散性,由于-NH2均匀地分散在聚合物表面,因而Au 纳米粒子也均匀地吸附到氨基酚醛树脂壳层上,Au 纳米粒子粒径约为5 nm,自身也未出现团聚现象,负载效果较好,这种简便的原位还原法还可以用来负载其他贵金属纳米粒子。

图5 Fe3O4@APF@Au 复合微球的TEM 照片Fig.5 TEM images of Fe3O4@APF@Au composite microspheres
为了进一步说明金纳米粒子已成功负载于Fe3O4@APF 复合微球表面,利用X 射线能谱分析仪分析了Fe3O4@APF@Au 磁性纳米复合催化剂中的元素,结果见图6 和表1。

图6 Fe3O4@APF@Au EDS 图Fig.6 EDS images of Fe3O4@APF@Au

表1 Fe3O4@APF@Au 能谱图的元素及含量Table 1 Elements and content in energy spectrum of Fe3O4@APF@Au
从图6 可以看到,合成的样品中除主要存在C,O 和Fe 的特征峰外,还存在Au 元素的峰位。其中C 元素主要来源于APF 树脂上的C,Fe 元素主要来自磁核Fe3O4,O 元素同时来源于磁核Fe3O4和APF 树脂壳。元素Au 的存在进一步说明贵金属已成功负载于Fe3O4@APF 壳层。表1 是对应EDS能谱图的各组成元素的具体含量,并且对所有经过分析的元素进行了归一化。
图7 是负载Au 纳米粒子后复合微球的XRD 图。Fe3O4@APF 负载Au 纳米粒子后,与原始Fe3O4@APF(图2)相比,在2θ 为38.3,44.5 和64.8°处出现了新的衍射峰,对比标准卡片JCPDS可知,该衍射峰分别对应Au 纳米粒子的(111),(200)和(220)晶面,说明Au 纳米粒子已成功负载到Fe3O4@APF 聚合物壳层上。

图7 Fe3O4@APF@Au 的XRD 图Fig.7 XRD patterns of Fe3O4@APF@Au

图8 加入硼氢化钠前后4-硝基苯酚的紫外吸收光谱Fig.8 Ultraviolet absorption spectra of 4-nitrophenol before and after adding sodium borohydride
2.3 复合催化剂Fe3O4@APF@Au 催化性能研究
氨基苯类化合物是化工生产中非常重要的一类中间体,将硝基苯类化合物还原反应成氨基苯类化合物是重要的有机转化反应之一[7]。在实验中选择4-硝基苯酚的还原反应作为探针[8-9],测试Fe3O4@APF@Au纳米复合材料的催化活性。
图8为未加催化剂的情况下,在4-硝基苯酚中加入硼氢化钠前后的紫外吸收光谱图。由图可知,在4-硝基苯酚中加入硼氢化钠后,4-硝基苯酚的紫外特征吸收峰发生了红移,特征峰从318.5 nm移到了401.5 nm,这是由于硼氢化钠溶液呈碱性,使呈弱酸性的4-硝基苯酚转化成了4-硝基苯酚钠。此外,在未加入催化剂情况下,即使加入过量的还原剂硼氢化钠,反应24 h,样品的紫外吸收光谱基本无变化。
当微量Fe3O4@APF@Au复合催化剂加入到上述体系中后(催化剂质量分数为0.05%),随着反应的进行,溶液由鹅黄色逐渐变为无色,取该反应过程中不同时间段的样品进行紫外光谱测试,结果见图9。由图可看出,4-硝基苯酚和硼氢化钠混合液在401.5 nm处的紫外特征吸收峰逐渐下降直到消失,这是由于加入催化剂后,4-硝基苯酚逐渐被硼氢化钠还原成了4-氨基苯酚,相应地在300 nm左右的紫外特征峰逐渐升高,表明在30 min内,4-硝基苯酚已经被完全还原。
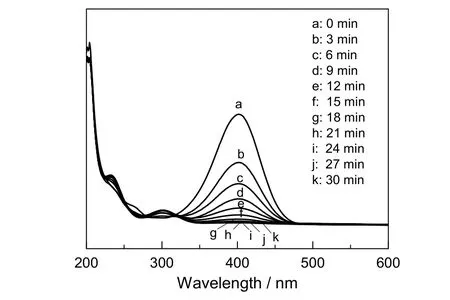
图9 Fe3O4@APF@Au纳米复合材料催化4-NP还原反应过程中样品的紫外吸收光谱Fig.9 Ultraviolet absorption spectra of samples during the Fe3O4@APF@Au nanocomposite catalyzed 4-nitrophenol reduction reaction
通过计算催化反应转化频率,可以精确标度Fe3O4@APF@Au 复合催化剂的催化性能。对于本实验中4-NP 的催化反应,由于硼氢化钠浓度远高于底物4-NP 的浓度(CNaBH4/C4-NP为400),所以可认为硼氢化钠在催化过程中浓度保持不变,因此4-NP 的还原过程可看做是催化底物的一级反应。因为不同时间下底物在401.5 nm 处的吸光度正比于当时底物的浓度,故选取图9 中各曲线在401.5 nm 处的吸光度值,以ln(Ct/C0)(Ct指代反应时间t 时催化底物的吸光度,C0指代催化底物的初始吸光度)对t作图,得到如图10 所示的线性关系图,表明反应符合准一级反应动力学,由反应速率方程ln(Ct/C0)=-kt计算得Fe3O4@APF@Au(0.05%)的一级反应速率常数(k)为8.52 h-1。进一步计算得知,本实验制备的复合催化剂Fe3O4@APF@Au 的TOF 值为3 187.7 h-1,表明Fe3O4@APF@Au 对4-NP 的还原反应具有非常好的催化活性。

图10 ln(Ct/C0)与t 的关系Fig.10 The relationship between ln (Ct/C0) and reaction time

图11 Fe3O4@APF@Au 纳米复合材料对4-NP 还原反应的循环催化性能Fig.11 Cyclic catalytic performance of Fe3O4@APF@Au nanocomposite for 4-nitrophenol reduction reaction
为了测试Fe3O4@APF@Au 纳米复合材料的稳定性,进行了循环催化性能测试,结果见图11。由图可知,在连续10 次循环催化实验中(每次催化5 min),4-硝基苯酚的转化率均达到93%,说明Fe3O4@APF@Au 纳米复合材料在经历10 次连续催化后催化活性无明显下降,证明其对4-硝基苯酚的还原反应有良好的循环催化稳定性。
2.4 复合催化剂Fe3O4@APF@Au 的磁性回收
循环再利用性能是工业化催化剂的关键指标之一,图12 展示了Fe3O4@APF@Au 在外加磁场下的行为。每次催化反应后,使用外加磁场可快速分离回收催化剂。图12(a)为在水溶液中超声分散好的Fe3O4@APF@Au,溶液均一稳定,当其被放在外加磁场下时,1 min 即可被完全分离[如图12(b)所示],经超声分散后又可恢复成图12(a)的状态,说明样品具有很好的磁响应性,可以通过外加磁场对其进行快速回收,节省了能源,增加了催化剂的使用效率。
3 结 论

图12 Fe3O4@APF@Au 纳米复合材料的磁性可控分离回收照片Fig.12 Photos of Fe3O4@APF@Au nanocomposite controlled magnetic separation and recovery a-no external magnetic field is applied; b-approximately 1 min after applying external magnetic field
核壳型磁性氨基酚醛树脂复合微球Fe3O4@APF 作为催化剂载体,氨基酚醛树脂壳层既保证了磁核不受外界环境的腐蚀又不影响磁性回收,贵金属Au 纳米粒子已负载于Fe3O4@APF 壳层中,负载均匀,分散性较好。以4-硝基苯酚的还原反应为探针,核壳型磁性复合催化剂Fe3O4@APF@Au 表现出了较高的催化活性和磁性可控分离回收效果,且经过10 次循环催化测试,依然保持较高的催化活性,具有很好的应用前景。
