功能化磁性纳米复合材料Fe3O4-mPD/SP吸附Cr(Ⅵ)研究
2020-05-15杨鑫宇吴杰张建庭吴纯鑫赵德明
杨鑫宇,吴杰,张建庭,吴纯鑫,赵德明
(浙江工业大学化学工程学院,浙江杭州310014)
引 言
重金属Cr(Ⅵ)具有高毒性、持久性、难降解等特点,它通过食物链进入生物体后与蛋白质、脂肪酸或氨基酸等结合,聚集到一定量后,严重危害人体和其他生物体健康[1-3]。针对水体Cr(Ⅵ)严重污染的现状,发展低耗能、高效率、适用范围广泛的Cr(Ⅵ)污染水体修复技术,已经变得越来越重要。
目前,国内外去除水体Cr(Ⅵ)的方法主要有化学沉淀法、离子交换法、生物法以及吸附法等[4]。吸附法以其高效、经济、简便等优点得到了广泛应用。传统的吸附剂都是利用吸附剂较大的比表面积及较高的表面能通过物理吸附去除水中的Cr(Ⅵ),这些材料大多存在选择性差、再生难、易产生二次污染等缺点。随着纳米技术的发展,纳米Fe3O4颗粒广泛应用于含Cr(Ⅵ)废水的去除[5-6],然而,在处理Cr(Ⅵ)废水过程中纳米Fe3O4颗粒也存在一些问题:暴露在空气中易被氧化,磁偶极相互作用使其易于团聚等[7-9]。所以,必须对纳米Fe3O4颗粒进行保护和改性修饰,在其表面引入化学稳定性强的官能团,如氨基(—NH2)、羧基(—COOH)、磺酸基(—SO3H)、羟基(—OH)等[10]。再配合使用超声波,超声波的空化作用、热效应和机械剪切作用能改善纳米粒子的矿物学特性,极大提高纳米颗粒的分散性,避免纳米颗粒发生团聚,提高反应活性[11]。目前单个官能团氨基修饰的磁性纳米Fe3O4用于吸附Cr(Ⅵ)的研究较多,如Huang 等[12]在纳米Fe3O4表面上先引入羧基,再通过碳二亚胺活化得到氨基修饰的磁性纳米材料Fe3O4@PAA-DETA,对Cr(Ⅵ)的饱和吸附量达到11.24 mg·g-1。Zhao 等[13]先反应得到环氧基功能化的纳米Fe3O4颗粒,再和乙二胺经开环反应得到氨基修饰的纳米Fe3O4复合材料EDA-NMPs,吸附量达到61.35 mg·g-1。这些对Cr(Ⅵ)的去除以物理吸附为主。Lv 等[14]采用一步法合成S-nZVI 材料,对Cr(Ⅵ)的吸附量达到64.10 mg·g-1,去除机制包括物理吸附、还原降解和共沉淀,具有良好的去除效果,但是极易发生团聚,限制了其在工程上的应用。
本研究在超声波辐照条件下,先制备出磁性纳米Fe3O4颗粒,再在超声波辐照下通过化学氧化法对其进行官能团修饰,制备得到富含氨基、亚氨基和磺酸基的磁性纳米复合材料Fe3O4-mPD/SP(50∶50)(间苯二胺单体(mPD)和2,5-二氨基苯磺酸单体(SP)以50∶50 摩尔比共聚包裹在Fe3O4纳米颗粒表面)作为废水中Cr(Ⅵ)的吸附剂,着重考察了溶液pH、吸附剂投加量、竞争性阴离子浓度、温度等因素对吸附效果的影响,建立了动力学模型,吸附等温线,计算出了热力学参数,并对其作用机理进行了探测,以期为实际应用提供基础参数。
1 实验部分
1.1 主要试剂
氯化铁(FeCl3·6H2O,≥99.0%,AR),国药集团化学试剂有限公司;七水合硫酸亚铁(FeSO4·7H2O,99.0-101.0%,AR),国药集团化学试剂有限公司;氨水(NH3·H2O,25%~28%,AR),衢州巨化试剂有限公司;间苯二胺(C6H8N2,99.5%,AR),麦克林试剂有限公司;2,5-二氨基苯磺酸(C6H8N2O3S,98%,AR),麦克林试剂有限公司;重铬酸钾(K2Cr2O7,≥99.8%,优级纯),阿拉丁试剂有限公司;二苯氨基脲(C13H14N4O,AR),麦克林试剂有限公司;丙酮(C3H6O,≥99.5%,AR),杭州双林化工试剂厂;过硫酸铵(H8N2O8S2,98.5%,AR),麦克林试剂有限公司;聚乙二醇(PEG-400,AR),阿拉丁试剂有限公司;氢氧化钠(NaOH,97%,CP),杭州萧山化学试剂厂;盐酸(HCl,36-38%,优级纯),国药集团化学试剂有限公司;硝酸(HNO3,65%~68%,AR),国药集团化学试剂有限公司;硫酸(H2SO4,98%,CP),衢州巨化试剂有限公司;磷酸(H3PO4,85%,AR),上海凌峰化学试剂有限公司;氮气(高纯氮),杭州今工特种气体公司。
1.2 超声波强化制备功能化磁性纳米复合材料
在超声波辐照下(40 kHz,150 W),向通N2保护的250 ml 三口烧瓶内按照摩尔比2∶3 分别加入FeSO4·7H2O 和FeCl3·6H2O,一定体积无氧去离子水混合均匀,滴加1 ml PEG-400作表面活性剂,100 ml恒压滴液漏斗缓慢滴入1.0 mol·L-1新鲜配制的氨水溶液至溶液pH=11,50℃水浴条件下搅拌反应1 h,搅拌速度450 r·min-1。反应完成后,无氧去离子水洗涤数次至中性。60℃真空干燥12 h,得磁性纳米Fe3O4颗粒。
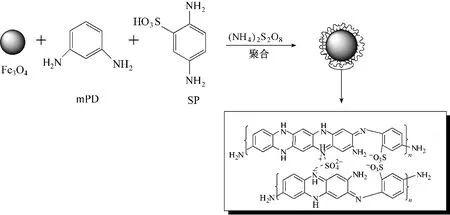
图1 磁性纳米Fe3O4颗粒的表面功能化Fig.1 Surface functionalization of magnetic Fe3O4 nanoparticles
采用化学氧化法一步合成氨基、亚氨基和磺酸基修饰的功能化磁性纳米复合材料。在超声波辐照下,称取0.5 g 上述步骤制备的磁性纳米Fe3O4、1 mmol 间苯二胺单体(mPD)和1 mmol 2,5-二氨基苯磺酸单体(SP)于250 ml 三口烧瓶,100 ml 无氧去离子水混合均匀,超声分散10 min,得a 溶液;称取2 mmol(NH4)2S2O8溶于10 ml 无氧去离子水,得b 溶液;将b 溶液以3 秒每滴的速度滴加到a 溶液,滴加结束,搅拌反应12 h,磁分离,无氧无离子水洗涤多次,60℃真空干燥12 h,制得超声波强化功能化磁性纳 米 复 合 材 料Fe3O4-mPD/SP(50∶50),反 应 式见图1。
1.3 样品表征
粒子的形貌采用日本电子株式会社的JEM-2100F 型透射电子显微镜表征;化学成分用Thermo Electron Corporation 公司的Nicolet 6700 红外光谱仪检测;磁性能采用PPMS-9T 型综合物性测试系统在298 K,-20~20 kOe(1 Oe=79.5775 A·m-1)时测量其磁滞回线;采用美国TA Q5000IR热重分析仪对粒子进行热重分析, 吹扫气体为高纯氮气,流速20 ml·min-1;晶体衍射采用PNAlytical 公司的X’Pert PRO型X 射线衍射仪,扫描范围为20°~80°;使用ASAR2020,Micromeritics 比表面分析仪以氮吸附法测定样品的比表面积。
1.4 复合材料的耐酸碱性研究
将50 mg Fe3O4、超声波强化制得的Fe3O4-mPD/SP(50∶50)分别加入50 ml 自来水、0.1 mol·L-1HCl、1 mol·L-1HCl、0.1 mol·L-1NaOH、1 mol·L-1NaOH 溶液中,常温下浸泡12 h 后磁分离,取上清液测定其总铁离子和mPD/SP 聚合物的溶解量。总铁含量采用原子吸收分光光度计测定,溶解的mPD/SP 聚合物采用总有机碳分析仪测定其溶解的有机碳。
1.5 吸附实验
用K2Cr2O7配制基于Cr(Ⅵ)的初始浓度为100 mg·L-1的模拟废水,稀HCl 和NaOH 用于调节溶液pH,取50 ml Cr(Ⅵ)溶液于锥形瓶,Fe3O4-mPD/SP(50∶50)投加量1 g·L-1,全温培养振荡器振荡频率180 r·min-1,吸附温度为303 K,恒温振荡吸附540 min,吸附完成后磁分离,取上清液测Cr(Ⅵ)浓度。采用二苯碳酰二肼(DPCI)分光光度法于540 nm 处测定吸光度。根据式(1)计算吸附容量。

式中,q 为吸附容量,mg·g-1;C0为Cr(Ⅵ)初始浓度,mg·L-1,Ce为吸附达到平衡时溶液中Cr(Ⅵ)浓度,mg·L-1;V为溶液体积,ml;m为吸附剂用量,mg。
2 结果与讨论
2.1 样品的表征
不同方法制备的Fe3O4-mPD/SP(50∶50)纳米复合材料形貌表征见图2。由图可知,不同方法制备的样品均为球形颗粒,粒径分布均匀,约50 nm。加入超声波后,样品分散性得到了显著提高。深色部分为Fe3O4,周围浅色部分为mPD/SP聚合物包覆层,表明富含氨基、亚氨基和磺酸基官能团的mPD/SP聚合物已成功包裹在Fe3O4表面。
图3 分别为纳米Fe3O4颗粒、普通方法和超声波强化制备的Fe3O4-mPD/SP(50∶50)XRD 谱图,谱图中均出现了Fe3O4的7 个典型特征峰(30.1°,35.5°,43.3°,53.4°,57.2°,62.6°和74.8°),分别对应于Fe3O4不同的晶面(220),(311),(400),(422),(511),(440)和(533),无其他杂质峰,表明制得的Fe3O4纳米颗粒较纯,且表面功能化修饰时没有改变Fe3O4颗粒的晶体构型,不会引起Fe3O4晶相变化。

图2 不同方法制备的Fe3O4-mPD/SP(50∶50)纳米复合材料的TEM图Fig.2 TEM images of Fe3O4-mPD/SP(50∶50)nanocomposites synthesized by different methods
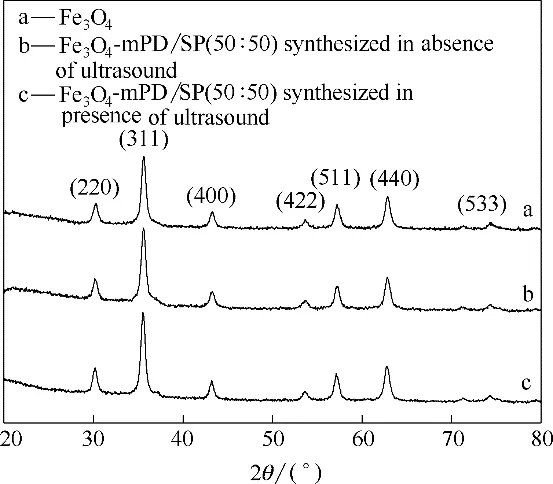
图3 磁性纳米Fe3O4颗粒和不同方法制备的Fe3O4-mPD/SP(50∶50)的XRD谱图Fig.3 XRD patterns of magnetic Fe3O4 nanoparticles and Fe3O4-mPD/SP(50∶50)synthesized by different methods
图4 曲线a 为纳米Fe3O4颗粒的红外光谱图,596.8 cm-1和1635 cm-1处是Fe—O 键振动吸收峰。3400 cm-1左右的峰可能为表面—OH 或水分吸收峰。曲线b、c 分别为超声波强化和普通方法制备的功能化纳米复合材料红外谱图,b和c在1635 cm-1处的峰明显强于a 纯Fe3O4,可能是因为b 和c 的N—H键和Fe—O 键振动吸收峰重合导致,因此,b 和c 在3400 cm-1处较大较宽的峰为N—H 键特征吸收峰,表明存在大量的氨基和亚氨基,这些氨基和亚氨基来源于Fe3O4-mPD/SP(50∶50)表面的mPD/SP 共聚物。在1025 cm-1和1072 cm-1处的吸收峰分别是SO键的非对称和对称伸缩振动,SO 键来自SP 单 元中—SO3-基团。超声波强化制备的Fe3O4-mPD/SP(50∶50)官能团吸收峰强度明显强于普通方法制备的Fe3O4-mPD/SP(50∶50)。可能是因为超声波强化制备的Fe3O4-mPD/SP(50∶50)表面的mPD/SP共聚物多于普通方法制备的Fe3O4-mPD/SP(50∶50)导致。
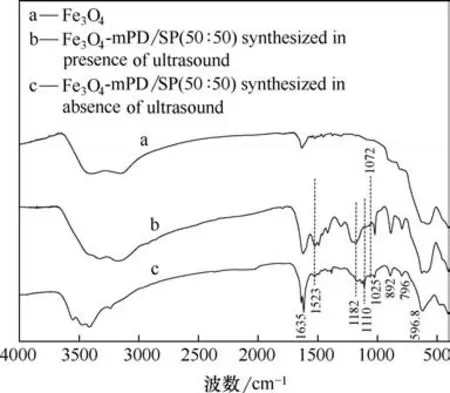
图4 磁性纳米Fe3O4颗粒和不同方法制备的Fe3O4-mPD/SP(50∶50)的红外谱图Fig.4 IR spectra of magnetic Fe3O4 nanoparticles and Fe3O4-mPD/SP(50∶50)synthesized by different methods
图5 分别为纳米Fe3O4颗粒、普通方法和超声波强化制备的Fe3O4-mPD/SP(50∶50)热重分析谱图。小于200℃之前的失重,是由于表面吸附的游离水分蒸发所致,700℃后,质量基本稳定,不再变化,200~700℃之间有失重主要因为Fe3O4-mPD/SP(50∶50)表面的mPD/SP 共聚物层的微量分解。从图中可以看出,随着温度的不断升高,纳米Fe3O4颗粒质量基本保持不变,超声波强化制备的Fe3O4-mPD/SP(50∶50)质量变化明显大于普通方法制备的Fe3O4-mPD/SP(50∶50),表明超声波强化制备的Fe3O4-mPD/SP(50∶50)表面的mPD/SP 共聚物多于普通法制备的Fe3O4-mPD/SP(50∶50),计算得出超声波强化复合材料Fe3O4-mPD/SP(50∶50)共聚物层mPD/SP 百分含量约为26.36%,Fe3O4百分含量约为69.98%,普通法制得的Fe3O4-mPD/SP(50∶50)共聚物层mPD/SP 百分含量约为9.23%,Fe3O4百分含量约为85.63%。
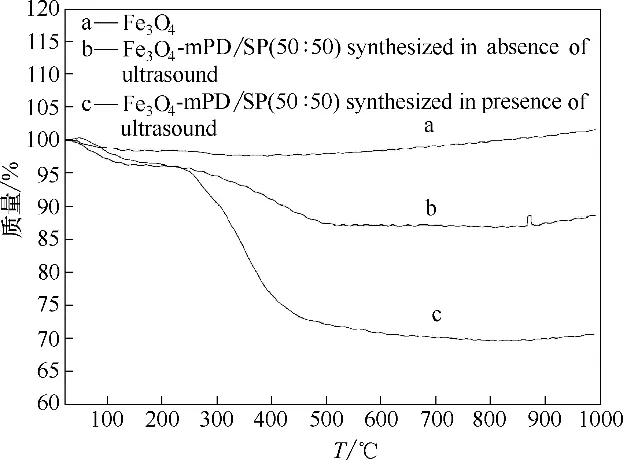
图5 磁性纳米Fe3O4颗粒和不同方法制备的Fe3O4-mPD/SP(50∶50)的热重谱图Fig.5 Thermogravimetric spectra of magnetic Fe3O4 nanoparticles and Fe3O4-mPD/SP(50∶50)synthesized by different methods
图6 为磁性纳米颗粒的磁滞回线,曲线a、b 和c分别为纳米Fe3O4颗粒、普通方法和超声波强化制备的Fe3O4-mPD/SP(50∶50),饱 和 磁 强 度 分 别 为57.64、52.17 和45.02 emu·g-1。Fe3O4在进行功能化修饰后,其饱和磁强度有所降低,相比于普通方法制备的磁性纳米材料,加入超声波强化后,饱和磁强度更小。可能是由于加入超声后共聚物mPD/SP的量增多导致。图中可观察到在外加磁场下,仅需30 s,Fe3O4-mPD/SP(50∶50)就可从水溶液分离,可见超声波强化制备的Fe3O4-mPD/SP(50∶50)磁响应强度良好,能够实现有效磁分离。
BET 表征结果表明:超声波强化制得的Fe3O4-mPD/SP(50∶50)的比表面积(105.75 m2·g-1)明显大于普通方法制得的Fe3O4-mPD/SP(50∶50)比表面积(84.93 m2·g-1)。究其原因是制备过程加入超声波后,强化界面间的传递和反应过程,有利于反应表面更新,超声波所具有的高温分解、分散和剪切破碎作用,能改善晶体构型、形态,提高其分散性。同时避免颗粒团聚,提高其比表面积,从而制得粒径更小、比表面积更大和化学反应活性更高的纳米复合材料。
2.2 复合材料的耐酸碱性研究
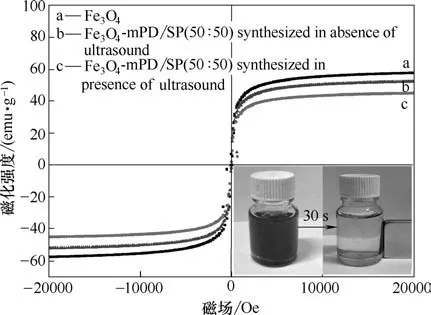
图6 磁性纳米Fe3O4颗粒和不同方法制备的Fe3O4-mPD/SP(50∶50)的磁滞回线谱图Fig.6 Hysteresis loop spectrum of magnetic Fe3O4 nanoparticles and Fe3O4-mPD/SP(50∶50)synthesized by different methods
由表1 可知,纯Fe3O4颗粒在各类水溶液中均有铁溶解,在1 mol·L-1HCl 溶液中溶解量最大,达到28.542 mg·L-1。经过功能化修饰后,Fe3O4纳米颗粒表面的mPD/SP 共聚物层加强了对Fe3O4颗粒的保护,铁离子的溶出量明显减小,在1 mol·L-1HCl溶液中,超声波强化制得的Fe3O4-mPD/SP(50∶50)的铁离子的溶出量只有1.729 mg·L-1。本实验通过测定溶液中的总有机碳,来判断mPD/SP 的溶解量,在1 mol·L-1HCl和1 mol·L-1NaOH 溶液中也只有微量的mPD/SP 被溶解。因此,超声波强化制得的Fe3O4-mPD/SP(50∶50)具有较好的耐酸碱性。
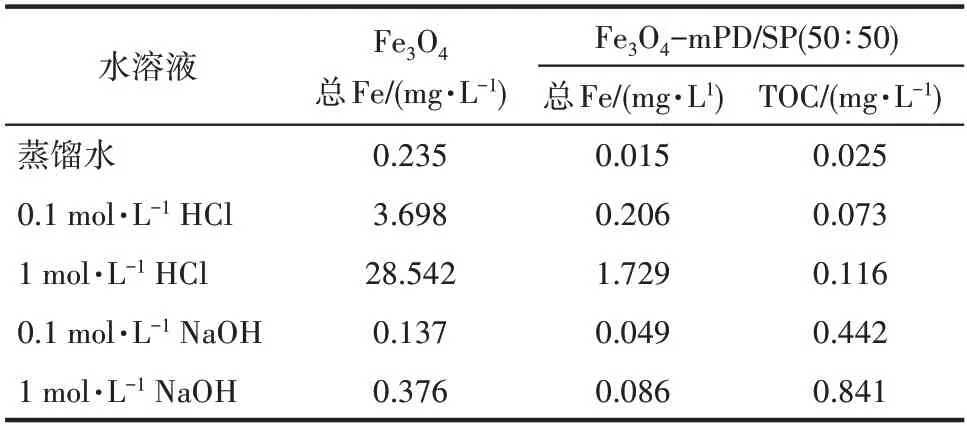
表1 纳米Fe3O4和超声波强化制得的Fe3O4-mPD/SP(50∶50)在不同溶液中总铁和mPD/SP溶解量Table 1 Total iron and mPD/SP solubility of nano-Fe3O4 particles and ultrasonic-enhanced Fe3O4-mPD/SP(50∶50)in different solutions
2.3 不同方法制备的Fe3O4-mPD/SP(50∶50)对Cr(Ⅵ)吸附效果对比
在pH=2,Cr(Ⅵ)初始浓度为100 mg·L-1,吸附剂投加量1 g·L-1,振荡频率180 r·min-1,T=30℃条件下进行吸附实验,结果见图7。
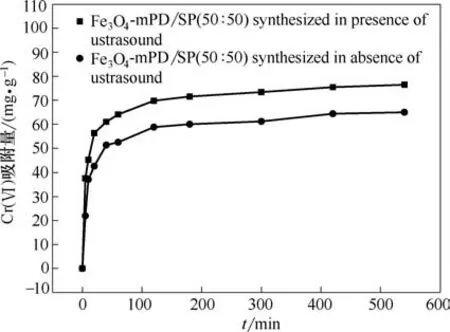
图7 不同方法制备的Fe3O4-mPD/SP(50∶50)对Cr(Ⅵ)吸附效果Fig.7 Effect of Fe3O4-mPD/SP(50∶50)synthesized by different methods on adsorption of Cr(Ⅵ)
由图7 可以看出,当利用超声波强化制备的Fe3O4-mPD/SP(50∶50)对Cr(Ⅵ)进行吸附时,540 min后吸附量达到76.434 mg·g-1;而在同样条件下用普通方法制备的Fe3O4-mPD/SP(50∶50)对Cr(Ⅵ)进行吸附时,540 min 后吸附量只有64.941 mg·g-1,说明在对Cr(Ⅵ)进行吸附过程中,超声波强化制备的Fe3O4-mPD/SP(50∶50)具有更优异的性能。同时与文献[3,12-15]已报道的几种吸附剂对Cr(Ⅵ)的吸附效果进行了比较,结果见表2。因此,在后续吸附实验中均使用超声波强化制备的Fe3O4-mPD/SP(50∶50)纳米复合材料。
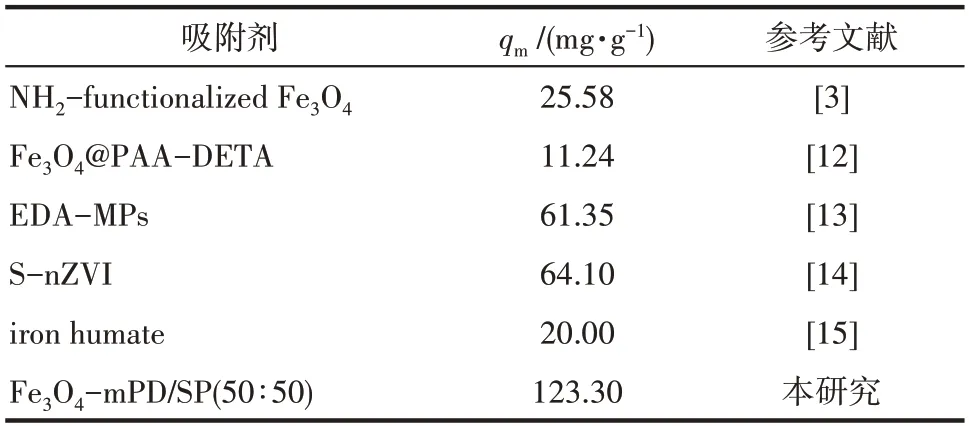
表2 不同吸附剂对Cr(Ⅵ)的吸附容量比较Table 2 Comparison of adsorption capacity of different adsorbents to Cr(Ⅵ)
2.4 不同mPD/SP 单体比例对Cr(Ⅵ)吸附能力对比
图8 为pH=2,Cr(Ⅵ)初始浓度100 mg·L-1,吸附剂投加量1 g·L-1,振荡频率180 r·min-1,T=30℃条件下,不同mPD/SP 单体比例(100∶0、95∶5、90∶10、75∶25、50∶50、25∶75、10∶90、0∶100,摩 尔 比)修 饰 的Fe3O4-mPD/SP 对Cr(Ⅵ)吸附能力对比。随着SP 单体含量从0增加到100%,Fe3O4-mPD/SP对Cr(Ⅵ)的吸附容量先增加后减小,当SP 单体含量为50%时,即Fe3O4-mPD/SP(50∶50)时,对Cr(Ⅵ)的吸附容量最大,达到76.434 mg·g-1,单独用mPD 或SP 修饰的Fe3O4-mPD/SP(100∶0)、Fe3O4-mPD/SP(0∶100)对Cr(Ⅵ)的吸附容量均小于Fe3O4-mPD/SP(50∶50)对C(rⅥ)的吸附容量,这可能是因为在SP单体含量为50%时,共聚物链上形成了活性氨基、亚氨基和磺酸基的最优组合结构,该最优组合结构有利于Cr(Ⅵ)与N—/N+H—/—NH2/—SO3H 官能团之间发生有效反应。图8 中,随着SP 单体含量从50%增加到100%,Fe3O4-mPD/SP 对Cr(Ⅵ)的吸附容量逐渐降低,这是因为Cr(Ⅵ)主要是以阴离子形态的HCrO4-存在于溶液中,HCrO4-与—SO3-之间会产生静电斥力,导致吸附容量随着SP单体含量继续增大而逐渐降低。因而本实验选用Fe3O4-mPD/SP(50∶50)作为后续研究中对Cr(Ⅵ)的吸附材料。
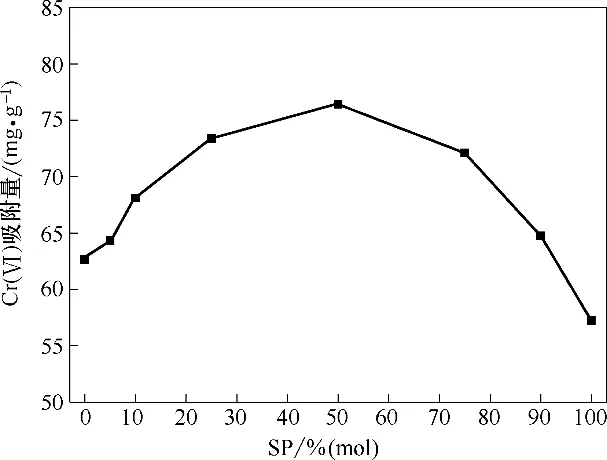
图8 不同mPD/SP单体比例的Fe3O4-mPD/SP吸附能力比较Fig.8 Comparison of adsorption capacity of Fe3O4-mPD/SP with different ratio of mPD/SP monomers
如图8 所示,Fe3O4-mPD/SP(100∶0)和Fe3O4-mPD/SP(0∶100)吸附Cr(Ⅵ)的吸附容量分别为62.672 mg·g-1和57.234 mg·g-1,如果包裹在Fe3O4表面的是mPD-mPD 和SP-SP 聚合物的简单混合物,Fe3O4-mPD/SP(50∶50)对Cr(Ⅵ)吸附量应该为59.953 mg·g-1(62.672×50%+57.234×50%=59.953 mg·g-1),实际Fe3O4-mPD/SP(50∶50)对Cr(Ⅵ)吸附量为76.434 mg·g-1,说明包裹在Fe3O4表面的的聚合物为mPD/SP 的共聚物,而不是mPD-mPD 和SP-SP聚合物的简单混合物。
2.5 初始pH值对吸附的影响
当Cr(Ⅵ)初始浓度为100 mg·L-1,吸附剂投加量1 g·L-1,振荡频率180 r·min-1,T=30℃时,考察pH在1~8范围内变化对Cr(Ⅵ)吸附量的影响。
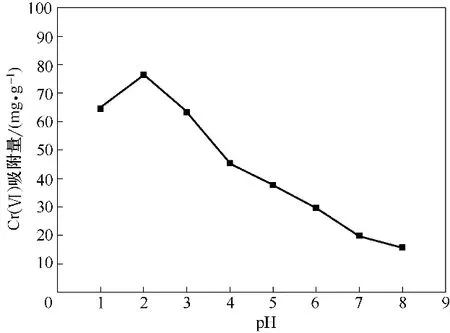
图9 pH对吸附效果的影响Fig.9 Effect of pH on adsorption
由图9可知,溶液pH对吸附效果影响较大。pH由1 增大至2 时,吸附量呈上升趋势,pH=2 时,吸附量达到最大,之后随着pH 继续增大,Fe3O4-mPD/SP(50∶50)对Cr(Ⅵ)吸附量直线减小。溶液pH 影响Fe3O4-mPD/SP(50∶50)去除Cr(Ⅵ)的因素主要有以下三个方面:(1)—NH2/—NH—/—SO3H 官能团存在形态受溶液pH 影响较大,在较低pH 条件下,—NH2/—NH—易与溶液中的H+质子化形成—NH3+/—NH2+—而带上正电,进而与带负电的产生强的静电引力吸附溶液中的Cr(Ⅵ);(2)溶液pH 对Cr(Ⅵ)在水溶液中的存在形式影响较大,当pH<2 时主要以H2CrO4和HCrO4-的形式存在;pH在2~6.5之间时以HCrO4-为主要存在形式;pH >6.5 时主要以CrO42-存在于溶液。不同形式存在的Cr(Ⅵ)被吸附时需要的吸附自由能不同。HCrO4-吸附自由能低于CrO42-的吸附自由能,HCrO4-比CrO42-更容易被吸附,随着溶液pH 增大,以HCrO4-为主要存在形式的Cr(Ⅵ)逐渐转变成CrO42-,不利于Cr(Ⅵ)被吸附;(3)随着pH 的升高,溶液中OH-浓度增大,OH-会与Cr(Ⅵ)竞争吸附点位,降低吸附剂对Cr(Ⅵ)的吸附容量。而pH 小于2 时,Fe3O4-mPD/SP(50∶50)对Cr(Ⅵ)吸附量减小,是由于此时Cr(Ⅵ)在溶液中以H2CrO4为主要存在形式,导致Cr(Ⅵ)总阴离子浓度减小,吸附作用力减弱,不利于Cr(Ⅵ)的吸附。以上分析表明,Fe3O4-mPD/SP(50∶50)吸附Cr(Ⅵ)时存在静电引力吸附机理,且此后均在pH=2条件下进行吸附实验。
2.6 吸附剂投加量对Cr(Ⅵ)吸附性能的影响
在pH=2,Cr(Ⅵ)初始浓度100 mg·L-1,振荡频率180 r·min-1,T=30℃条件下,考察吸附剂投加量对吸附的影响,结果见图10。由图可知,随着吸附剂投加量增加,对Cr(Ⅵ)去除率呈上升趋势,这是因为吸附剂投加量增加导致吸附活性位点数目也随之增加,在Cr(Ⅵ)水溶液浓度不变的情况下,Cr(Ⅵ)被吸附的量也会增加,因此Cr(Ⅵ)去除率增大。但是,单位质量吸附剂对Cr(Ⅵ)的吸附容量呈现下降趋势。因为新吸附剂的加入,使平衡的流动相浓度减少,原来吸附剂表面吸附的Cr(Ⅵ)会解吸,单位质量吸附剂的吸附容量减小,吸附剂利用率变低。探究吸附剂对Cr(Ⅵ)的去除率以及吸附剂单位吸附量来确定最佳吸附剂投加量,随着吸附剂投加量的增加,单位质量吸附剂吸附量是减小的,而对Cr(Ⅵ)的去除率是增大的,二者有一个交点,选择交点附近的吸附剂用量最优。因此,选用吸附剂投加量为1 g·L-1。
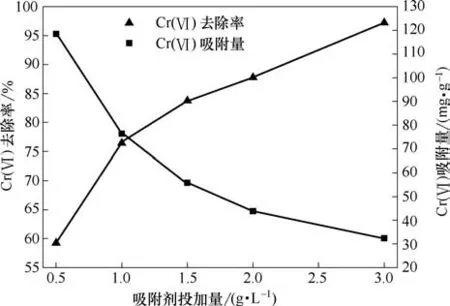
图10 吸附剂投加量对吸附的影响Fig.10 Effect of adsorbent dosage on adsorption
2.7 竞争性离子的影响
在pH=2,Cr(Ⅵ)初始浓度100 mg·L-1,吸附剂投加量1 g·L-1,振荡频率180 r·min-1,T=30℃条件下,考察竞争性阴离子(SO42-,Cl-和NO3-)浓度为0、0.01、0.05、0.1、0.2 mol·L-1时对吸附的影响,结果如图11 所示,从图中可看出,竞争性阴离子(SO42-,Cl-和NO3-)的存在并未对Fe3O4-mPD/SP(50∶50)吸附Cr(Ⅵ)造成显著影响。随着Cl-、NO3-和浓度的增大,Fe3O4-mPD/SP(50∶50)吸附Cr(Ⅵ)的吸附量略有下降。对吸附的影响比Cl-和大。因为pH=2 时,Cr(Ⅵ)以HCrO4-形式存在于水溶液,竞争性阴离子对吸附的影响是由于产生了竞争吸附。不同的阴离子与Fe3O4-mPD/SP(50∶50)相互作用能力不同,所带电荷量高的与吸附剂作用力较强,SO42-带两个电荷,作用力强于Cl-和NO3-,被吸附后会占据更多的吸附点位。因此,竞争性阴离子SO42-对吸附的影响强于Cl-和NO3-。
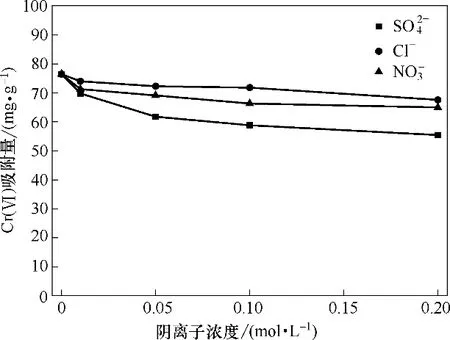
图11 竞争性离子对吸附的影响Fig.11 Effect of competitive ions on adsorption
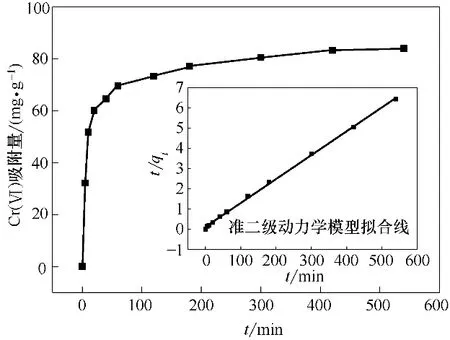
图12 吸附量随时间变化的曲线及吸附动力学模型拟合Fig.12 Curve of adsorption capacity with time and fitting of adsorption kinetics model
2.8 吸附动力学研究
在pH=2,Cr(Ⅵ)初始浓度100 mg·L-1,吸附剂投加量1 g·L-1,振荡频率180 r·min-1,T=30℃条件下,定时5、10、20、40、60、120、180、320、540 min 取样测定溶液中Cr(Ⅵ)浓度,结果如图12 所示。Fe3O4-mPD/SP(50∶50)对Cr(Ⅵ)的吸附在前30 min吸附速度很快,5 h 后基本达到吸附平衡。Fe3O4-mPD/SP(50∶50)的吸附过程包括两个步骤[16]:最先的快速步骤和后续的慢速控制步骤,最先的快速步骤有可能是Cr(Ⅵ)与Fe3O4-mPD/SP(50∶50)表面官能团发生的还原反应和物理吸附作用引起的,而后续的 慢 速 控 制 步 骤 是HCrO4-向mPD/SP 共 聚 物 里 面的内部扩散以及HCrO4-和—SO3-产生的静电斥力控制的。为进一步研究吸附过程,采用准一级、准二级动力学模型[17]和内扩散模型[18]对实验数据进行拟合。
拟合结果见表3,准一级动力学、准二级动力学和内扩散方程的相关系数R2分别为0.932、1.000 和0.748。准二级动力学方程拟合线能很好地与数据点重合,更好地描述Fe3O4-mPD/SP(50∶50)对Cr(Ⅵ)的吸附过程。由于准二级动力学方程是基于吸附剂和吸附质之间的化学吸附为整个吸附过程的速度控制步骤的假设,因此可以认为吸附机制主要包括还原反应和Cr(Ⅵ)与官能团之间的络合吸附,且由准二级动力学方程计算所得的平衡吸附量为77.157 mg·g-1,与实验测定值76.434 mg·g-1非常相近。
2.9 吸附等温线
在pH=2,Cr(Ⅵ)初始浓度100 mg·L-1,吸附剂投加量1 g·L-1,振荡频率180 r·min-1,T=30℃时,考察Fe3O4-mPD/SP(50∶50)对Cr(Ⅵ)的等温吸附情况。图13 是Fe3O4-mPD/SP(50∶50)对Cr(Ⅵ)的吸附等温线。由图可知,Fe3O4-mPD/SP(50∶50)对Cr(Ⅵ)的吸附量q 随平衡浓度Ce的增加而增大,然后接近某一固定值,即饱和吸附量qm。对Cr(Ⅵ)等温吸附数据分 别 采 用Langmuir[式(2)]、Freundlich[式(3)]和Temkin[式(4)]方程进行拟合[19]。
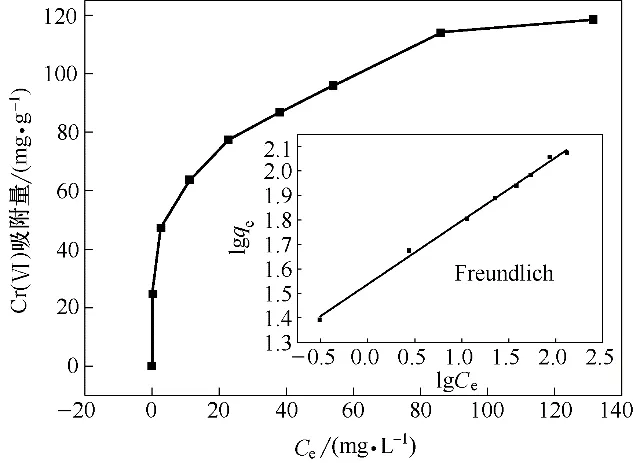
图13 吸附等温线及吸附等温线拟合Fig.13 Adsorption isotherm and adsorption isotherm fitting
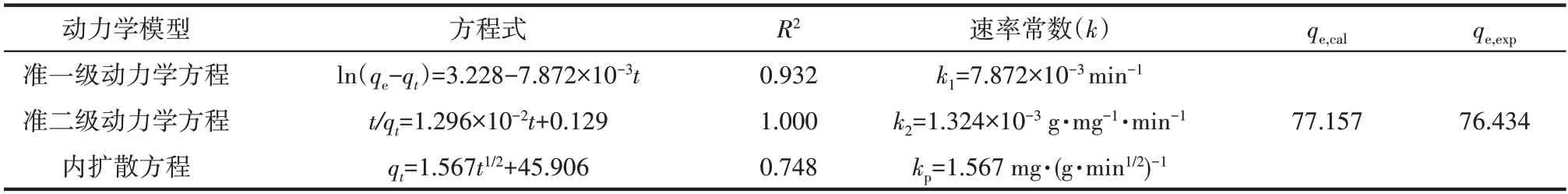
表3 Fe3O4-mPD/SP(50∶50)对Cr(Ⅵ)吸附动力学拟合方程Table 3 Fitting equation of adsorption kinetics of Cr(Ⅵ)by Fe3O4-mPD/SP(50∶50)

表4 Fe3O4-mPD/SP(50∶50)对Cr(Ⅵ)的吸附等温线拟合参数Table 4 Fitting parameters of adsorption isotherms of Cr(Ⅵ)by Fe3O4-mPD/SP(50∶50)
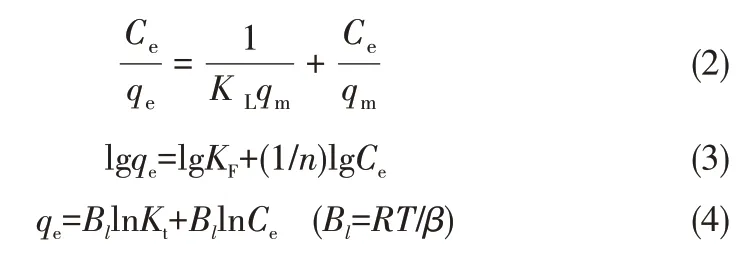
式中,qe为平衡吸附量,mg·g-1;qm为饱和吸附量,mg·g-1;Ce为吸附达到平衡时浓度,mg·L-1;KL为Langmuir 吸附速率常数;KF为Freundlich 吸附系数,Kt、β为Temkin吸附等温式常数。
相关参数如表4所示。通过比较几种模型的拟合相关系数R2,Freundlich 模型的线性相关性更好(R2=0.996)。因此Freundlich 模型能更好地描述Fe3O4-mPD/SP(50∶50)对Cr(Ⅵ)的吸附过程。KF是用来描述吸附剂吸附性能的参数,KF越大说明吸附剂的吸附性能越好,当0.1<1/n<0.5 时,表明Cr(Ⅵ)易于被Fe3O4-mPD/SP(50∶50)吸附,当1/n>2 时表明Cr(Ⅵ)难以被Fe3O4-mPD/SP(50∶50)吸附,本实验测得KF=34.464 mg1-(1/n)·L1/n·g-1,n=3.861,表明吸附剂性能较好,吸附过程容易进行(1<n<10)。
2.10 吸附热力学
在pH=2,Cr(Ⅵ)初始浓度100 mg·L-1,吸附剂投加量1 g·L-1,振荡频率180 r·min-1条件下,将锥形瓶分别放置在20、30、40 和50℃的恒温振荡器中振荡吸附540 min,反应结束磁分离,取上清液测定Cr(Ⅵ)。结果如图14 所示,随着吸附温度的升高,Fe3O4-mPD/SP(50∶50)对废水中Cr(Ⅵ)的吸附量有所增大,这表明温度的升高有利于吸附的进行。计算反应的Gibbs自由能ΔG0,熵变ΔS0及焓变ΔH0,结果如表5所示[20]。
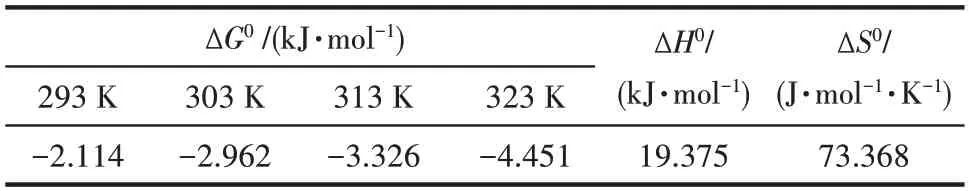
表5 Fe3O4-mPD/SP(50∶50)吸附Cr(Ⅵ)热力学常数Table 5 Thermodynamic constants of adsorption of Cr(Ⅵ)by Fe3O4-mPD/SP(50∶50)
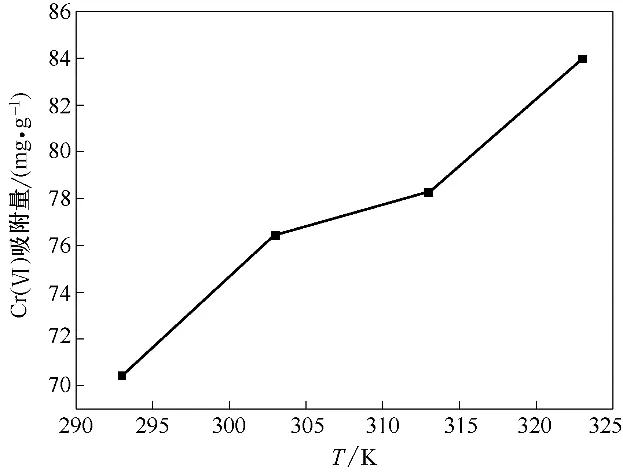
图14 温度对吸附的影响Fig.14 Effect of temperature on adsorption
由表5 可见,不同吸附温度下,吸附反应的ΔG0均小于零,说明该吸附反应能自发进行。熵变ΔS0为73.368 J·mol-1·K-1,说 明Fe3O4-mPD/SP(50∶50)对Cr(Ⅵ)的吸附是一个熵增大的反应。吸附焓变ΔH0为19.375 kJ·mol-1,该吸附反应是吸热过程,升高反应温度,有利于吸附反应的进行,Fe3O4-mPD/SP(50∶50)对Cr(Ⅵ)的吸附容量增大,与实验结果一致。
2.11 重复性实验
由表1可知,本文制得的Fe3O4-mPD/SP(50∶50)具有较好的耐酸耐碱性,本实验釆用0.1 mol·L-1的硝酸为解吸剂解吸Fe3O4-mPD/SP(50∶50)。图15为Fe3O4-mPD/SP(50∶50)重复利用5 次对应的吸附量,由图可知,随着重复利用次数的增多,吸附剂的吸附量有所下降,原因可能有以下三点:①在数次的解析过程中,Fe3O4-mPD/SP(50∶50)的聚合物包裹层mPD/SP 有少部分溶解在稀硝酸溶液中,造成氨基、亚氨基、磺酸基活性基团的数量减少,导致吸附量下降;②解析不彻底,Fe3O4-mPD/SP(50∶50)上面仍有部分Cr(Ⅵ)未被洗脱出来,仍占据吸附位点;③吸附过程中,苯型仲胺和Cr(Ⅵ)之间发生了氧化还原反应,吸附剂表面mPD/SP 分子链上的苯型仲胺被氧化为醌型亚胺,重复利用数次后,吸附剂氧化还原能力会减弱,影响吸附效果。经过5 次吸附-脱附-再生后,其对Cr(Ⅵ)的吸附容量有所下降。
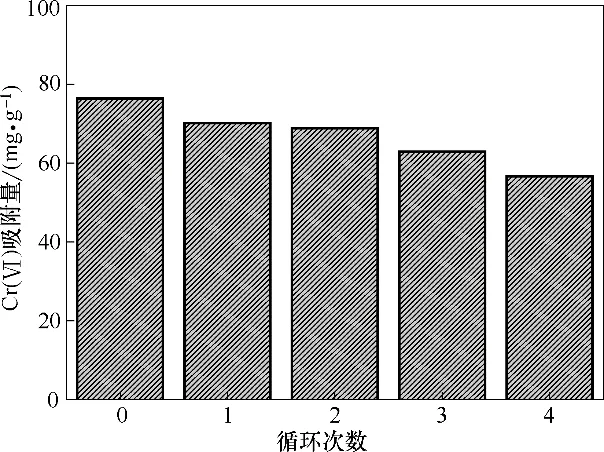
图15 循环使用次数对吸附量的影响Fig.15 Effect of recycling times on adsorption capacity
3 吸附机理探究
图16 为Fe3O4-mPD/SP(50∶50)吸附前后XPS 全扫描谱图,由图可知,吸附后XPS 全扫描谱图在结合能为580 eV 左右处出现了Cr 2p 新峰,表明Cr 已经成功被吸附[21]。图17 为Fe3O4-mPD/SP(50∶50)吸附Cr(Ⅵ)后的单扫描Cr 2p 谱图,由图可知,Cr 2p3/2和2p1/2 吸收峰分别出现在结合能为573 和582.85 eV 的位置,表明吸附结束后,吸附剂上Cr(Ⅲ)和Cr(Ⅵ)同时存在,其中Cr(Ⅲ)是由共聚物mPD/SP 上具有还原性的苯型仲胺通过氧化还原反应将Cr(Ⅵ)还原成的Cr(Ⅲ)。由2.2 节可知,在Fe3O4上包裹mPD/SP 共聚物后,只有微量Fe溶出,因此Cr(Ⅵ)被Fe(Ⅱ)还原忽略不计。
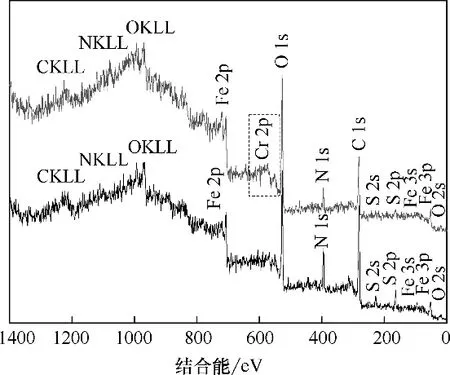
图16 Fe3O4-mPD/SP(50∶50)吸附Cr(Ⅵ)前后的XPS全扫描谱图Fig.16 XPS full scan spectra before and after adsorption of Cr(Ⅵ)by Fe3O4-mPD/SP(50∶50)
测量了吸附前后溶液pH 的变化。吸附Cr(Ⅵ)后,溶液的pH 从2.0 增加到2.71,是由于mPD/SP 分子链中的—NH2/—NH—和溶液中的H+发生质子化反应,消耗了游离H+,虽然质子化的氨基、亚氨基仍有可能与Cr(Ⅲ)发生离子交换使H+被释放到溶液中。但是消耗H+的速度大于离子交换释放出H+的速度,因此溶液pH 增大。由此可推出Fe3O4-mPD/SP(50∶50)吸附Cr(Ⅵ)的过程中存在氧化还原、静电吸附和离子交换吸附机理。
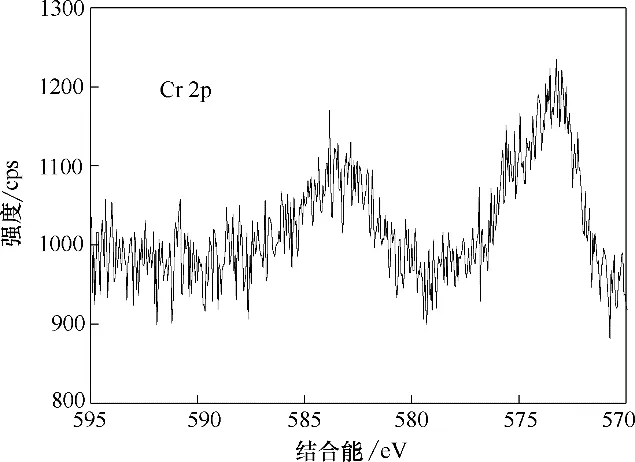
图17 Fe3O4-mPD/SP(50∶50)吸附Cr(Ⅵ)后的单扫描Cr 2p谱图Fig.17 Single scan Cr 2p spectrum after adsorption of Cr(Ⅵ)by Fe3O4-mPD/SP(50∶50)
图18 为Fe3O4-mPD/SP(50∶50)吸附前后红外光谱图,吸附后,1182 cm-1处吸收峰强度有所减小,此处吸收峰为mPD 苯环和SP 苯环中的C—N 键拉伸振动。可知,共聚物上的—N /—NH2/—NH—功能团可与Cr(Ⅲ)形成稳定的络合物而使Cr(Ⅲ)被吸附。1621 cm-1和1524 cm-1分别对应于醌型环和苯环的伸缩振动,吸附后,1621 cm-1处吸收峰强度与1524 cm-1处吸收峰强度比值增大,部分苯环通过氧化还原反应被Cr(Ⅵ)氧化变成醌型环[22]。吸附前后,Fe3O4-mPD/SP(50∶50)峰型基本没有变化,但是各特征吸收峰强度均有不同程度变小。表明吸附前后,吸附剂自身组成、结构没有变化,但是吸附剂表面活性官能团有所消耗。
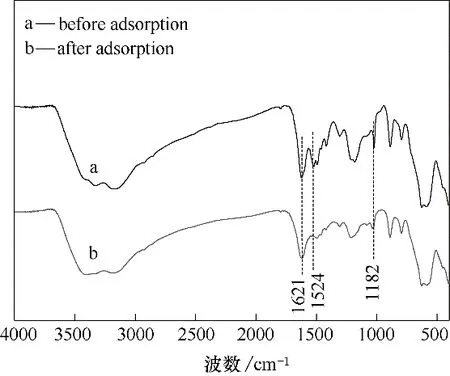
图18 Fe3O4-mPD/SP吸附Cr(Ⅵ)前后的红外光谱图Fig.18 Infrared spectra before and after adsorption of Cr(Ⅵ)by Fe3O4-mPD/SP
通过以上探究分析,Fe3O4-mPD/SP(50∶50)对Cr(Ⅵ)的吸附历程[23]可能为:
(1)mPD/SP 共聚物的—NH2/—NH—官能团发生质子化反应而带正电,同时溶液中存在大量Cr(Ⅵ)阴离子,通过静电引力作用Cr(Ⅵ)阴离子大量被吸附。同时部分Cr(Ⅵ)经过物理吸附附着于吸附剂表面;
(2)被吸附的Cr(Ⅵ)阴离子接近吸附剂表面mPD/SP 分子链上苯型仲胺,苯型仲胺和Cr(Ⅵ)发生氧化还原反应,苯型仲胺被氧化为醌型亚胺,Cr(Ⅵ)被还原为Cr(Ⅲ)
(3)Cr(Ⅵ)或者还原得到的Cr(Ⅲ)被亚胺基团鳌合或以物理吸附的方式吸附在共聚物mPD/SP表面。
机理见图19。
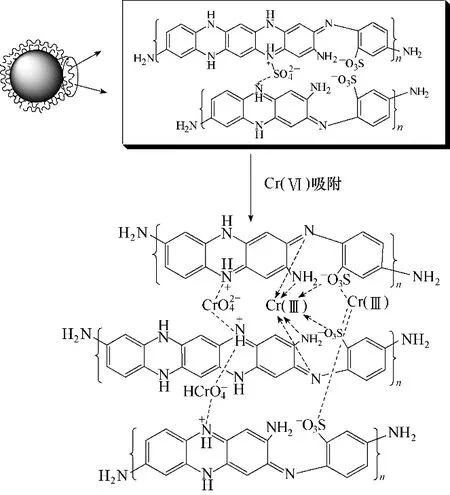
图19 功能化磁性纳米复合材料Fe3O4-mPD/SP(50∶50)吸附Cr(Ⅵ)机理Fig.19 Adsorption mechanism of Cr(Ⅵ)adsorbed by Fe3O4-mPD/SP(50∶50)
4 结 论
(1)本文在超声波辐照下制备了功能化磁性纳米复合材料Fe3O4-mPD/SP(50∶50),并采用TEM、XRD、IR、TGA、VSM 及BET 对其进行了表征。考察了溶液pH、吸附剂投加量、竞争性阴离子浓度、温度等对吸附性能的影响。
(2)利用Langmuir、Freundlich 和Temkin 方程对Cr(Ⅵ)等温吸附数据进行拟合,发现Freundlich 模型能更好地描述Cr(Ⅵ)在Fe3O4-mPD/SP(50∶50)上的吸附行为,且吸附过程容易进行。
(3)Fe3O4-mPD/SP(50∶50)吸附Cr(Ⅵ)的热力学研究表明:Fe3O4-mPD/SP(50∶50)对Cr(Ⅵ)的吸附为自发过程;吸附反应为吸热过程,升温有利于吸附的进行。经过5 次吸附-脱附-再生后,超声波强化功能化磁性纳米复合材料Fe3O4-mPD/SP(50∶50)对Cr(Ⅵ)的吸附容量有所下降。
(4)推测出超声波强化功能化磁性纳米复合材料Fe3O4-mPD/SP(50∶50)对Cr(Ⅵ)的吸附过程中存在静电吸附、氧化还原和离子交换吸附机理。
