新型小分子抗凝血药物的合成、作用机制及构效关系研究进展
2020-05-13李梦瑶蔡志强
李梦瑶, 蔡志强, 侯 玲*, 李 帅
(1. 沈阳工业大学 石油化工学院 辽宁省芳烃下游精细化工工程技术研究中心,辽宁 辽阳 111003;2.山东省药学科学院 化学药物重点实验室,山东 济南 250101)
目前只有较少数量的新型小分子抗凝血药物在临床上获得应用,如作用于Ⅱa因子靶点的直接凝血酶抑制剂达比加群酯以及直接Xa因子抑制剂利伐沙班、阿哌沙班、依度沙班等[1-3]。静脉血栓栓塞对人类健康的威胁十分严重,因此治疗静脉血栓的新型抗凝药物一直是药物研发的一大热点。与大分子药物及华法林相比,新型小分子口服抗凝血药物具有诸多优点:抗凝作用不依赖于抗凝血酶,具有良好的剂效关系,口服起效快、半衰期短,与食物和药物之间基本不发生相互作用,口服无需监测常规凝血指标,可减少因用药不当造成的药物疗效下降或出血等不良事件,可预测的药动学特性、更好的药效以及更高的安全性[4],以Ⅱa因子和Xa因子为靶点的小分子抗凝血药物的开发成功,为血栓患者带来了新的希望。
Scheme 1
本文对已上市的Ⅱa因子和Xa因子新型小分子抗凝血药物的合成、作用机制及构效关系研究进展进行综述,并对该类药物的发展趋势和前景进行展望。
凝血酶Ⅱa因子是丝氨酸蛋白酶,也是凝血级联反应中的关键酶。凝血酶是一种“胰蛋白酶样”丝氨酸蛋白酶蛋白质,在凝血作用第一级联反应中,凝血酶原即凝血因子Ⅱ被蛋白水解性切除生成凝血酶,凝血酶促使可溶性的纤维蛋白原转化为纤维蛋白单体,最终纤维蛋白单体相互聚合形成不溶于水的交联纤维蛋白多聚体(不溶性的纤维蛋白),同时激活凝血因子V、 Ⅷ、 Ⅺ,并继续刺激产生凝血酶。各种凝血因子与凝血酶协同作用,促进发生凝血级联反应,从而达到止血的目的。此外,Ⅱa因子还可活化血小板,从而启动凝血过程,是抗凝药开发的重要靶点。随着对Ⅱa因子三维结构的了解,陆续开发了一系列与凝血酶特异性结合的直接凝血酶抑制剂[1-4]。
1.1 希美加群(Ximelagatran)
2004年6月于德国上市的希美加群是第一个进入临床应用并作用于Ⅱa因子的新型口服直接凝血酶抑制剂,其可通过乙基的脱烷基化和羟基的脱羟基化两个途径,经小肠吸收后迅速转化为具有抗凝活性的美拉加群(Melagatran)[5-8]。美拉加群是一种类似纤维蛋白肽A的二肽,能与凝血酶活化位点结合使其失活,从而抑制血小板活化,减少纤溶时间,发挥抗凝作用[6-9]。
Antonsson等[10]报道了希美加群的合成路线(Scheme 1),此路线以N-(叔丁基氧基羰基)-(R)-苯甘氨酸(1)为原料,经还原、缩合、水解等4步反应得到化合物6,化合物6在去苄氧羰基的合成过程中采用Pd/C催化的方法得到化合物7,然后与化合物8进行酯交换反应得到得到化合物9,再用三氟乙酸脱Boc基团得到化合物10,化合物10与2-(三氟甲基磺酰氧基)乙酸乙酯发生缩合反应,得到希美加群的前体药物12,化合物12在乙醇中与羟胺反应生成目标化合物希美加群(13)。该合成路线反应条件温和,前药(12)具有药用活性,产品收率高,适合工业化大量生产。Sorbera等[11]在中间体的基础上,经过盐酸去Boc得到化合物14,化合物14再与化合物15发生胺酯交换反应得到化合物16,最后经Pd/C还原得到希美加群(13)。该合成路线反应条件苛刻,试剂昂贵,不适合工业化大量生产。
通过对希美加群构效关系的研究[7-9,12-13]发现,美拉加群是D-Phe-Pro-Arg三肽序列的类似物,对血浆蛋白的结合亲和力较低,药物相互作用的可能性也较低,且未涉及细胞色素P450酶的临床相关药物相互作用。将D-Phe-Pro-Arg三肽用苯脒基代替精氨酸,吖丁啶-2-羧酸代替脯氨酸,D-型环己基甘氨酸代替D-苯丙氨酸,即为美拉加群的结构。由于美拉加群其含有羧基、仲胺基和脒基,亲水性高,口服利用度低。将美拉加群的羧基酯化,脒基成肟后便转化为可口服的希美加群。希美加群可迅速抑制凝血酶地产生[5],其作用优于依诺肝素。希美加群的生物利用度为18%~24%,半衰期为4~5 h,约80%经由肾脏排出[6-7,14]。希美加群主要不良反应是引起转氨酶增高,发生率约为6%。该药由于无需凝血功能监测以及剂量调整,因此比早期的口服抗凝药物(如维生素K拮抗剂等)具有明显的优势,曾被认为是理想的口服抗凝药。但在安全性评估时发现该药有6%的患者存在肝毒性,容易引起肝功能损害,甚至引发急性肝功能衰竭而导致死亡。因此,该药于2004年获得美国FDA批准,于2006年被停止使用且退出市场[11,14-16]。但希美加群的获批上市推动了全球口服抗凝药的发展,其退市并未削弱世界制药巨头公司放弃对这一领域的开发,在某种程度上以此为鉴,激发了药物学家开发该领域药物的极大兴趣。
1.2 达比加群酯(Dabigatran Etexilate)
2010年10月,德国勃林格殷格翰公司研发的作用于Ⅱa因子的新一代口服直接凝血酶抑制剂(DTIs)抗凝药物,达比加群酯(泰毕全)获美国FDA批准上市[17-23]。达比加群酯适用于卒中、短暂性脑缺血发作或全身性栓塞的患者。达比加群酯在肝脏代谢中经过口服转化为具有直接抗凝血活性的达比加群,其可阻止纤维蛋白原裂解为纤维蛋白阻止血栓的形成并且和凝血酶的纤维蛋白特异位点结合[12-27]。
李泽标等[28],Norbert等[29],Zerban等[30]分别报道了达比加群酯的合成路线(Scheme 2)。李泽标等以对氨基苯甲腈(17)为原料,分别经环合、水解、酰化、缩合等反应生成达比加群酯(33)。该制备达比加群酯的方法原料便宜易得,操作简单,收率高,纯度高,适合大规模工业化生产。Norbert等报道的合成路线是以3-硝基-4-氯苯甲酸(24)为起始原料,共经过7步反应得到达比加群酯(33),该路线反应条件温和,操作简单;但反应步骤较多,合成繁琐,原料不易获得,且总收率低,使该制备路线的成本较高,不能满足工业化生产。Zerban等报道制备达比加群酯路线的方法是将化合物29和34在CDI作用下发生缩合反应,再经催化氢化反应得到化合物32,化合物32与氯甲酸正己酯(22)进行酰化反应后生成达比加群酯(33)。该路线中的中间体提纯方法复杂,且需要柱层析纯化,因此不利于进行大规模生产。

Scheme 2
通过研究达比加群的构效关系[13-14,27]发现,达比加群的羧基与靶点的Leu99和IIe174残基结合,脒基与靶点的Asp189、 Gly216和Trp215残基相连;将分子中吡啶环替换为苯环对其活性几乎没有影响,小的亲脂性官能团可取代与脒基相连的苯环,整个分子的空间骨架与靶点可以完美的键合。但是,分子中脒基的存在使药物地吸收及生物利用度明显降低,为克服达比加群脒基存在造成的生物利用度不高的缺点,可以对其脒基进行结构修饰和改造,使其达到更好的透膜吸收效果,具有较好的成药性。达比加群的清除率不依赖于细胞色素P450系统,且不影响从肝脏CYP2C9及CYP3A4酶系统代谢药物的活性[12],与其他药物间相互作用较少,安全性较高[14,18,21,27]。服用达比加群酯1.5 h血浆药物浓度可达到峰值,24 h后血浆浓度降低低,平均血浆浓度随剂量增加而增加,达比加群酯的稳定半衰期为12~17 h,生物利用度为7%[16,18,21,26],未吸收的达比加群酯主要经粪便排出[12,26,31-33]。ECT检测在临床相关药物浓度范围内与药物血浆浓度呈线性关系,且有足够的灵敏度和准确性[33-35]。新一代口服凝血酶抑制剂达比加群酯,不仅具有希美加群的药效优点,同时无明显肝功能损害。因此对达比加群酯及其系列产品的合成与开发将拥有广阔的应用前景。
Xa因子是维生素K依赖的糖基化丝氨酸蛋白酶,处于内源性凝血和外源性凝血通路的中心位置[2],每分子的Xa因子可活化产生约1000个分子的凝血酶[3,36],分子结构包括4部分[37-39]:S1、 S2、 S3、 S4。其中S1是Xa因子的特异性结合口袋,也是决定底物特异性的主要因素之一;S4是Xa因子的芳香性口袋,是发挥药效作用的位点,Xa因子抑制剂在结合位点上所采用的构象呈L型,能较好与其靶点的S1、S4口袋相互作用,其抑制剂种类按照是否依赖于AT-Ⅲ因子可分为直接抑制剂与间接抑制剂[3,27,32-44]。间接Xa因子抑制剂需要AT-Ⅲ因子作为辅助因子,不能抑制凝血酶原酶复合物结合的Xa因子;直接Xa因子抑制剂直接作用于Xa因子分子的活性中心,既抑制血浆中游离的Xa因子,也能抑制被凝血酶原酶复合物结合的Xa因子。因此,开发直接Xa因子抑制剂为抗凝药物的发展提供了新的研究方向。
2.1 利伐沙班(Rivaroxaban)
强生公司研发的利伐沙班于2011年7月获美国FDA批准上市,是全球第一个上市的口服Xa因子直接抑制剂,其治疗效果高于依诺肝素的新型口服抗凝药物,且能预防静脉血栓的产生[18-19]。
Mathlas等[44]报道了以4-(4-氨基苯基)-3-吗啉酮(36)为原料与化合物37反应得化合物38,化合物38在N,N′-羰基二咪唑(CDI)作用下生成化合物39,化合物39在甲胺盐酸盐作用下生成化合物40,化合物40与5-氯噻吩-2-酰氯(41)在碳酸钠作用下得到利伐沙班42(Scheme 3)。此反应条件温和,适合工业化放大生产。侯俊凯等[45]报道利伐沙班的制备方法是以(S)-4-氯-3-羟基丁腈(44)为原料,经邻苯二甲酰亚胺钾盐取代、腈基水解、Hofmann重排、偶联、肼解、酰胺化等反应得到利伐沙班(42)。在该合成路线中,避免使用贵金属催化剂及污染较大的试剂,使整个合成过程经济环保、易处理、收率和纯度较高、生产成本低、适合工业化生产。Susanne[46]和Masse等[47]报道了以(S)-3-氯-1,2-丙二醇(50)为原料,在碳酸钾作用下环合得化合物51,化合物51与邻苯二甲酰亚胺(52)反应得化合物53,化合物53与4-(4-氨基苯基)-3-吗啉酮反应得化合物54,化合物54在CDI作用下得化合物40,化合物40和5-氯噻吩-2-酰氯(41)在吡啶和四氢呋喃作用下生成利伐沙班(42)。该路线中所需原料价格昂贵,且来源有限,反应过程中生成杂质较多,不易于工业化生产。

Scheme 3
由利伐沙班的口服生物利用度(50%~55%)、半衰期(4~9 h)、达峰时间(2~4 h)可知,很少与食物发生相互作用[11-12,16,48-49],在使用过程中有66%的药物经肾脏代谢,其余经胆汁代谢[1,49],因此肾功能不全的患者亦可安全有效地使用该药。利伐沙班通过细胞色素P450系统进行肝脏清除,即CYP3A4、CYP2J2和CYP机制在肝脏中代谢[16,11,48,50]。4项RECORD实验[26,32,51-52]表明:利伐沙班相对依诺肝素来说预防静脉血栓更有优势;但利伐沙班的大出血发生率明显高于依诺肝素。作为一种口服特异性的Xa因子抑制剂,利伐沙班能高选择性和竞争性地与Xa因子的活性位点结合,抑制游离和结合的Xa因子及凝血酶原活性。根据利伐沙班构效关系[49,53-55]分析可知,利伐沙班分子中噁唑烷酮上的羰基和噻吩环附近的NH与Gly219残基发生相互作用,从而形成两个分子间的氢键。在两个氢键的作用下,使噁唑烷酮母核成为吗啉酮环与S4口袋结合以及氯噻吩部分与S1口袋结合的核心部分。其中,噁唑烷酮环与苯环处于同一平面上,苯环与Trp215残基结合。在S1口袋中,噻吩环的氯原子与Tyr228残基相互作用,5-氯噻吩甲酰胺部分与Asp189结合,可提高其特异选择性。在保证其空间骨架结构不变的基础上,改变S1及S4口袋的官能团结构,为新药开发提供了一定的思路。
2.2 阿哌沙班(Apixaban)
百时美施贵宝、辉瑞公司联合研制的阿哌沙班是继达比加群酯和利伐沙班之后的第3种口服抗凝药,于2012年获美国FDA批准上市[56]。
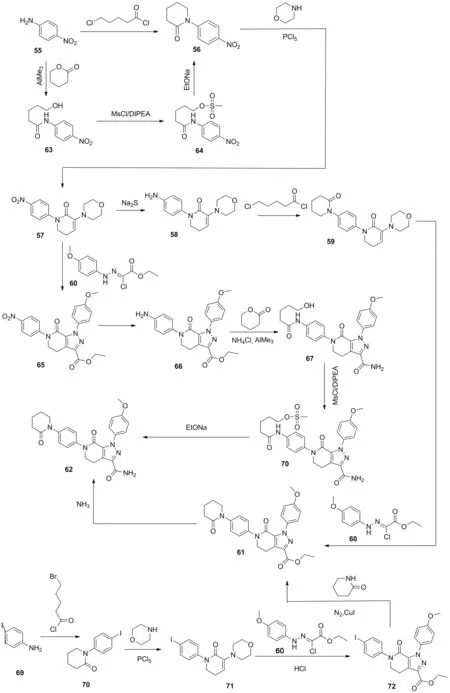
Scheme 4
冀亚飞等[57]报道了以对硝基苯胺(55)为起始原料(Scheme 4),在碱性条件下与5-氯戊酰氯经酰胺化-环合反应得到化合物56,化合物56再经过氯化、缩合、还原、酰胺化、环合和氨解等一系列反应,得到阿哌沙班(63)。该合成路线中以氢化钠作为酰胺化-环合步骤的缩合剂,生产成本高且操作危险性大,其中化合物57经硫化钠还原生成化合物58,此步反应易生成杂质,影响收率和纯度。赵金龙等[58]同样以对硝基苯胺(55)和δ-戊内酯为起始原料,在AlMe3作用下进行酰胺化、甲磺酰基取代、闭环反应得到化合物56。化合物57经过环合、硝基还原得到化合物66,化合物66再与合成化合物63的相同条件生成化合物67,再经过取代、环合反应得到阿哌沙班(62)。该制备路线反应条件温和,操作简单,产物收率高,纯度高,适合工业化大量生产。GANT等[59]报道了以对碘苯胺(69)为起始原料,与5-溴戊酰氯反应,依次经过酰胺化-环合、氯化、缩合、偶联和氨解等反应得到阿哌沙班(62)。由于该路线具有起始原料价格高,偶联反应条件苛刻,收率低等缺点,因此该路线不适合工业化生产。
阿哌沙班是一种比华法林能更有效地预防和治疗血栓的药物,此药的临床效果甚至比维生素K类产品更为出色。其可减少凝血酶原转化为凝血酶,从而发挥抗凝、抗栓的作用,继而减少由凝血酶引起的凝血系统中血小板的活化。阿哌沙班的IC50为0.51 nmol·L-1,药物口服生物利用度为60%~86%,稳定半衰期为10~14 h,血浆药物浓度达峰时间为用药后的1~2 h[2,8,14,18-19,27,51]。与利伐沙班相同,阿哌沙班通过CYP3A4和CYP机制在肝脏中代谢,其有25%的药物经肾脏排泄[2,11,16,50]。对阿哌沙班构效关系研究[42,56,60-64]可知,阿哌沙班结构中的酰胺键与P1口袋的Asp189残基相互作用,哌啶酮中的羰基与由Tyr99、Phe174和Trp215组成的S4芳香性口袋残基相互结合而发生作用。在研究中发现,对甲氧基苯基插入S1口袋,芳香酰胺一半处于疏水的S4口袋中。阿哌沙班在体内经代谢最终产物主要为O-去甲基阿哌沙班,其具有较好的生物活性,对其活性代谢产物的进一步开发,有望寻找到新的抗凝药物。
2.3 依度沙班(Edoxaban)
日本第一三共株式会社研制的小分子药物依度沙班于2015年1月获美国FDA批准上市,主要用于治疗减少非瓣膜性房颤卒中和全身性栓塞[11,51]。其为一种口服凝血Xa因子直接抑制剂,可抑制游离的Xa因子(Xa因子位于凝血级联的关键位置,处于凝血内外途径的汇合处),激活凝血酶原中的凝血酶。
NOGUCI等[65]报道了依度沙班的合成路线(Scheme 5),该路线以(1S,3R,4R)-4-叠氮基-3-[(叔丁氧羰基)氨基]-环己烷羧酸乙酯(73)为起始原料,经叠氮基还原,氨基脱保护与化合物77反应后,进行酯键的水解,再与二甲胺反应得到依度沙班(79)。该路线中化合物77较难制备,因此不适合工业化生产。Ohta[66]和尹新昊[67]报道了以(S)-3-环己烯-1-甲酸(80)为原料,经与碘的加成、酯化、亲核取代、钯碳催化还原、氨基保护、酯键水解、酰胺化等7步反应得到化合物87,再与化合物77反应得到化合物88,化合物88经过钯碳催化还原得到化合物89,最后与化合物90进行酰胺化反应得到依度沙班(79)。在该路线中用到难以合成的化合物77、 87以及90,该路线所需成本较高,操作复杂,因此不适合大量生产。李莹等[68]报道的依度沙班的制备方法通过调整反应顺序,减少了依度沙班制备路线中的缺点,在合成依度沙班(79)时,以叔丁基(1R,2S,5S)-2-氨基酸-5-(二甲氨基甲酰氯)环己基氨基甲酸甲酯(91)为起始原料,与化合物92进行胺酯交换反应得到化合物93,将化合物93去Boc反应得到化合物94,化合物94在1-羟基苯并三唑、1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐的作用下与化合物95反应得到依度沙班(79)。该合成方法改进了合成方案,提高了收率,减少了杂质的生成,合成路线简单,适合工业化大量生产。
依度沙班吸收快速,可直接与Xa因子活性位点结合且抑制活性不需监测凝血指标,其对凝血酶(IIa因子)的选择性比对Xa因子低10 000倍,并且对其它丝氨酸蛋白酶不产生相互作用。依度沙班是CYP3A4的底物,不是CYP1A2, CYP3A4和P-gp转运蛋白的抑制剂和诱导剂[64]。口服依度沙班的绝对生物利用度为67%,血浆峰浓度达峰时间为口服后1~5 h,血浆消除半衰期为6~11 h,35%经肾脏排出[40-49,64,69-70]。依度沙班构效关系研究[69-71]表明:依度沙班分子中的一个刚性环烷二胺结构不仅提高了该类结构的生物活性,而且极大的改善了其药代动力学性质。结构中5-氯-2-氨基吡啶通过二羰基连接到母核环烷二胺上,提高了其药效活性,并且使其与P1口袋很好的契合,同时保证了甲基哌嗪部分与P4口袋也能很好的键合。
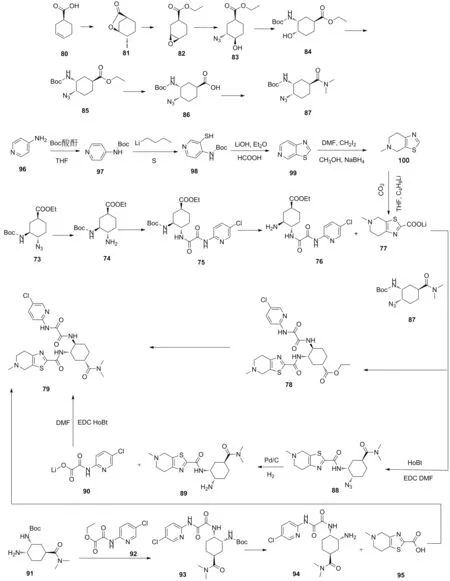
Scheme 5
2.4 贝曲沙班(Betrixaban)
默克公司与Portola公司共同研发的高选择性Xa因子直接抑制剂贝曲沙班于2017年6月获批上市,适应症为脑卒中及血栓栓塞[51-52,72]。贝曲沙班与其它口服Xa因子抑制剂相比,作用时间更持久,肾脏及肝脏清除率更低。
Kanter等[73]报道的贝曲沙班合成路线是以5-甲氧基-2-硝基苯甲酸(101)和2-氨基-5-氯吡啶(102)为原料(Scheme 6),在三氯氧磷的作用下进行酰胺化反应得到化合物103,化合物103经Pt/C催化加氢得到化合物104,再与4-氰基苯甲酰氯(105)作用生成化合物106,最后与二甲基酰胺锂在THF溶液中发生偶合反应得到贝曲沙班(107)。由于该路线在与二甲基酰胺锂反应中生成两种与贝曲沙班结构相似的杂质,从而影响贝曲沙班的收率和纯度。Pandey等[74]报道的制备贝曲沙班的合成路线是在Kanter等报道的合成路线基础上加以改进,从而避免了上述杂质的生成。在合成化合物104后,直接将4-氰基苯甲酰氯和二甲基酰胺锂加入反应体系中,生成含有脒基的化合物108,然后化合物104和化合物108在N,N-二甲基甲酰胺(DMF)和盐酸中反应得到贝曲沙班(107)。此路线避免了副产物的生成,提高了产物的收率和纯度,适合工业化生产。
贝曲沙班作为一种具有高选择性的直接Xa因子抑制剂,可通过抑制Xa因子活性,阻碍凝血酶的形成,继而阻碍纤维蛋白的形成,最终抑制血栓的形成和扩大[43]。贝曲沙班的IC50值为1.5 nmol·L-1,半衰期为19~20 h,生物利用度为47%,达峰时间为3~4 h,血浆浓度稳定,效应平稳,起效迅速,不依赖于细胞色素P450途径,无药物之间的相互作用[72,75]。药动学特征与药效效应可以预测,无需检测与调整剂量。根据贝曲沙班的构效关系[3,36-38]可知:贝曲沙班结构中的4-(N,N-二甲脒基)-苯甲酰胺基可与Xa因子的S4口袋完美结合,可有效地抑制Xa因子的活性;2-氨基-5-氯吡啶与S1位点键合;苯环上小的疏水性基团甲氧基可以提高抑制剂与Xa因子的结合亲和力。贝曲沙班通过与Xa因子的特异性结合,阻止了凝血酶原向凝血酶的转化,继而阻碍纤维蛋白的形成,最后有效地抑制了血栓的形成。由于贝曲沙班在急性病患静脉血栓的预防效果优于利伐沙班和阿哌沙班,故在此领域具有较好的优势,目前上市的沙班类品种未能达到该效果,因此其应用前景仍十分乐观。
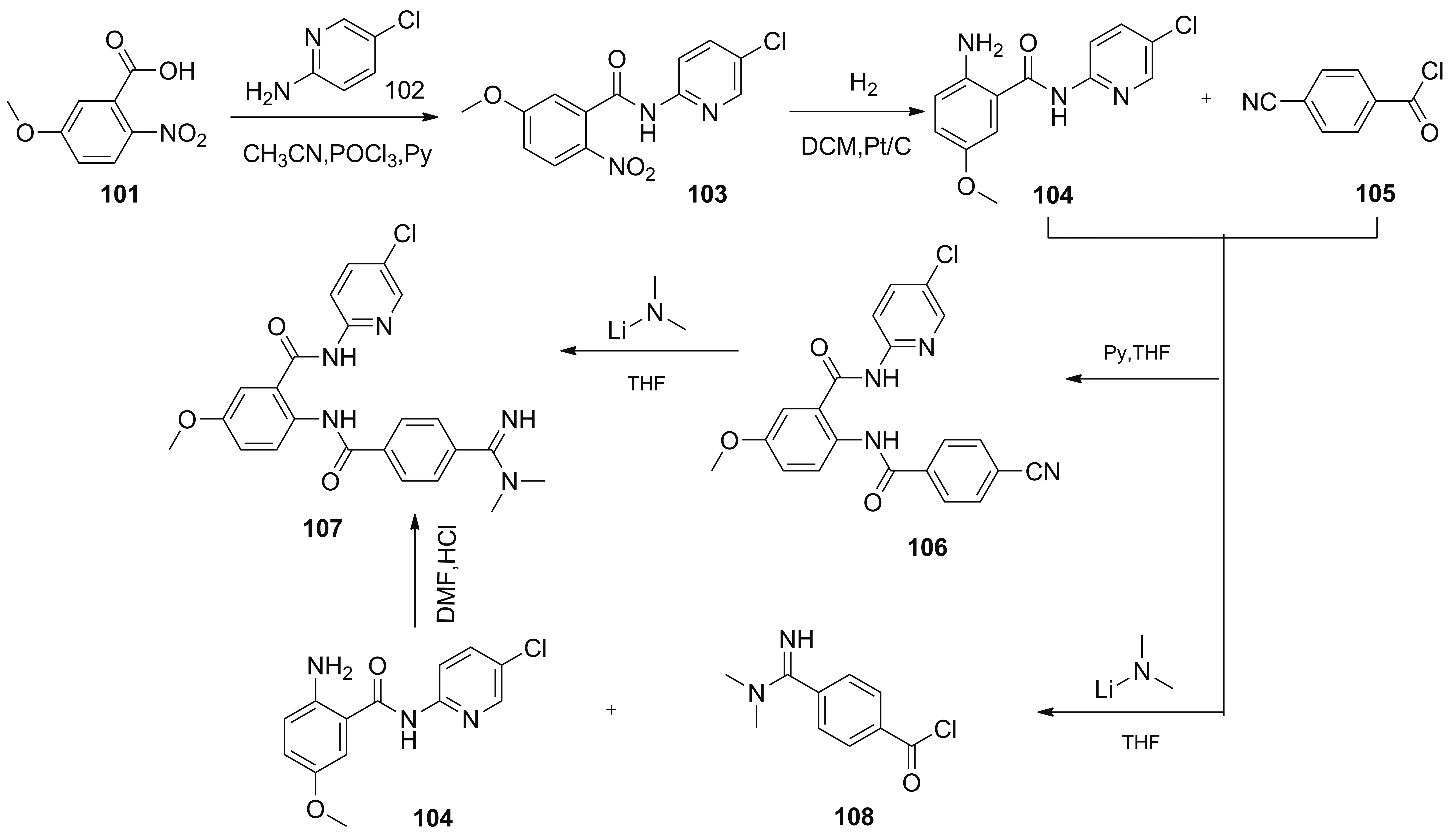
Scheme 6
综上所述,通过对最近开发的6个已上市的小分子抗凝血药物的合成、作用机制和构效关系分析发现,以Ⅱa因子和Xa因子为靶点的新型抗凝血小分子药物在抗凝领域起到了重要的作用,这些药物的研发为血栓治疗提供了更多的选择空间。对此类药物与作用靶点的深入细致分析将有助于抗血栓药物的进一步研发和临床应用。随着计算机辅助药物设计和化学合成新技术的有效结合,必将加速选择性更好、活性更强的新型抗凝血药物的发现和寻找。这将极大的推动抗凝药物的发展,并为发现低毒、高效的新型抗血栓药物提供新的策略和思路。
