2,5,6-位正交保护的5-氧-(喹啉-2-甲酰基)-β-D-半乳呋喃糖乙硫苷供体的合成
2020-05-13杨劲松
杨 攀, 杨劲松
(四川大学 华西药学院,四川 成都 610041)
多糖是真菌细胞壁的主要成分,也是真菌赖以生存和进化的重要物质[1]。作用于细胞壁多糖的抗真菌药物,如葡聚糖合成酶抑制剂、几丁质合成酶抑制剂、甘露聚糖合成抑制剂等[2],具有高效低毒的特点,有望成为临床上新一代抗真菌药物。2002年,Ahrazem等[3]从真菌Apodusdeciduus细胞壁中提取了水溶性多糖F1SS,并确证其结构是由甘露糖通过α-(1→6)糖苷键连接而成的聚合物,每个甘露糖3-位带有一个二糖片段,该二糖片段是由两个半乳呋喃糖通过α-(1→2)糖苷键连接而成的[4-5]。F1SS是组成多肽多聚糖的一部分,具有免疫原性,可参与细胞与细胞或细胞与宿主间的识别,但是有关F1SS的合成研究却尚未见报道,因此F1SS的合成具有十分重要的意义。合成该分子的主要挑战是两个连续的1,2-cis半乳呋喃糖苷键的构建。
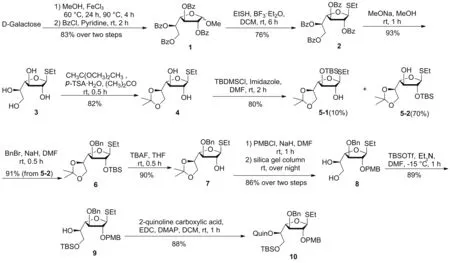
Scheme 1
1,2-cis半乳呋喃糖苷键的构建一直以来都是糖化学合成工作中所要攻克的重点和难点。因此,也吸引了很多研究者致力于该方向的研究:2001年,Kim课题组[6]设计了一种新型的糖基活化体,即将2-苄氧羰基苄基(BCB)作为甘露糖供体的离去基团,并将这种供体成功运用于1,2-cis甘露糖苷键的合成中。2006年,该课题组又将这个方法学应用到半乳呋喃糖中,高选择性地完成了抗肿瘤鞘糖脂agelagalastatin的全合成[7]。但是该方法在制备糖基活化体的过程中涉及剧毒物质氰化汞的使用,故其应用受到了一定程度的限制。2014年,Gola等[8]报道了3-位由Bz取代的半乳呋喃糖供体可通过远程参与控制糖苷键的立体构型,并用此方法合成了肺炎链球菌22F的荚膜多糖(CPS)的一个三糖片段。但该供体仅尝试了与鼠李糖受体的反应,其普适性有待验证。2013年,本课题组的刘强维等[9]将氢键介导的苷元传递策略(HAD)[10]用于构建1,2-cis阿拉伯呋喃糖苷键[11],用喹啉-2-甲酰基(Quin)保护阿拉伯呋喃糖供体的5-OH,在糖苷化过程中,保护基Quin上sp2杂化的氮原子与受体之间可以形成氢键,从而将受体束缚在糖环的β面,使得受体对端基中心的进攻以较高机率发生在β面,从而得到β构型占较大比例的糖苷化产物。该供体与多种受体反应均取得较好的选择性,同时,本课题组将此方法成功运用于结核分枝杆菌的脂阿拉伯甘露聚糖(LAM)的八糖片段合成中。之后,又将此方法不断地进行完善与拓展。2016年,本课题组高鹏程等[12]将该方法学扩展到1,2-cis半乳呋喃糖苷键的构建中,并用5-位由Quin保护的半乳呋喃糖供体高效地完成了从海绵中分离得到的鞘糖酯vesparioside B[13]的全合成。该方法具有选择性好,操作简便,安全性高的特点。
综上,为合成F1SS,本课题组设计了关键中间体10:以Quin保护化合物10的5-OH,用以控制1,2-cis半乳呋喃糖苷键的形成。基于F1SS的结构特点,采用临时保护基PMB和TBS分别保护化合物10的2-OH和6-OH,实现了2,5,6-位的正交保护。完成糖苷化反应之后,可以将5-位的Quin和6-位的TBS选择性脱除,换成位阻较小的保护基(如异亚丙基),而2-位的PMB脱除之后可作为受体,这些设计均有利于后续反应的进行。
本文以D-半乳糖为原料,参照文献[14-16]方法制得4,再通过TBS选择性保护其2-位制得5-2,用苄基(Bn)保护其3-位制得6[16],脱除TBS[6]之后用临时保护基PMB保护其2-位[8],并在洗脱过程中通过硅胶的弱酸性将其5,6-位的异亚丙基脱除,以较高的产率直接制得8,再于-15 ℃的低温条件下,通过分批加入TBSOTf,制得构型单一的产物9,最后用Quin保护其5-位制得化合物10[12](Scheme 1),总收率为18.6%,其结构经1H NMR,13C NMR和LC-MS(ESI)确证。
1 实验部分
1.1 仪器与试剂
Autopol Ⅵ型旋光仪;Varian Unit INOVA-400/54型核磁共振仪(CDCl3为溶剂,TMS为内标);Agilent 1260-6420型液相色谱质谱联用仪。
1~4[14-16], 6, 7, 10[6,8,16]按文献方法合成;其余所用试剂均为分析纯。
1.2 合成
(1) 5的合成
在干燥的圆底烧瓶中加入化合物4 2.90 g(11 mmol)和N,N-二甲基甲酰胺(DMF) 110 mL,冷却至0 ℃,加入咪唑2.24 g(33 mmol),搅拌15 min后,加入叔丁基二甲基氯硅烷(TBDMSCl) 2.42 g(16 mmol),升至室温反应2 h(TLC监测)。反应完全,用甲醇调至pH 7,加入乙酸乙酯200 mL,依次用水(3×30 mL)和饱和食盐水(30 mL)洗涤,有机层经无水硫酸钠干燥,减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:A=石油醚/乙酸乙酯=30/1,V/V)纯化得无色油状液体5-1 0.42 g,收率10%和5-2 2.91 g,收率70%。


(2) 6的合成[16]

(3) 7的合成[6]

(4) 8的合成
(5) 9的合成
在干燥的圆底烧瓶中加入化合物8 2.35 g(5 mmol)和DMF 50 mL,冷却至-15 ℃,缓慢滴加干燥的三乙胺(Et3N)0.83 mL(6mmol),滴毕,再缓慢滴加TBSOTf 1.49 mL(6 mmol)(分6批加入,每隔10 min加1次),加毕,反应至终点(TLC监测)。于-15 ℃加水终止反应,加入DCM 200 mL,依次用水(330 mL)和饱和食盐水(30 mL)洗涤,有机层用无水硫酸钠干燥,减压蒸除溶剂,残余物经硅胶柱层析(洗脱剂:A=12/1)纯化得无色油状液体9 2.64 g,收率89%,0.5, CHCl3);1H NMR(400 MHz, CDCl3)δ: 7.38~7.18(m, 7H), 6.95~6.80(m, 2H), 5.37(d,J=2.3 Hz, 1H), 4.61~4.47(m, 3H), 4.42(d,J=11.4 Hz, 1H), 4.23 (dd,J=6.6 Hz, 2.9 Hz, 1H), 4.10 (dd,J=6.6 Hz, 2.8 Hz, 1H), 3.96(t,J=2.6 Hz, 1H), 3.81(s, 3H), 3.78(s, 1H), 3.68~3.59(m, 2H), 2.76~2.58(m, 2H), 2.45( br s, 1H), 1.30(t,J=7.4 Hz, 3H), 0.90(s, 9H), 0.06(s, 6H);13C NMR(100 MHz, CDCl3)δ: 159.55, 137.82, 129.83, 129.38, 128.51, 127.91, 113.97, 87.86, 87.73, 83.62, 80.30, 72.36, 71.65, 71.17, 64.31, 55.38, 25.97, 25.48, 18.38, 14.98, -5.28, -5.32; LC-MS(ESI)m/z: Calcd for C29H44O6SSi{[M+Na]+}571.252 1, found 571.2522。
(6) 10的合成

2 结果与讨论
2.1 5的合成
在化合物5的合成过程中,文献[17]采用叔丁基二苯基氯硅烷(TBDPSCl)与化合物4在咪唑催化下反应,对2-位进行选择性保护,选择性为5/1(2-位/3-位),参照文献[6]把TBDPSCl换成了位阻较小的TBDMSCl,在保证收率的情况下将选择性提高到了7/1。
2.2 8的合成
在化合物8的合成过程中,本课题组曾参照文献[18]的方法,在65 ℃下用醋酸/水(4/1, V/V)的条件脱除异亚丙基,结果发现酸性及加热条件会导致PMB脱落,极大地降低了收率。尝试降低温度或醋酸的比例,但均未取得好的效果。在多次实验过程中,发现用硅胶柱纯化产物会使得异亚丙基有部分脱落,于是尝试延长产物在硅胶柱上的时间,上样后过夜再洗脱,发现异亚丙基可以完全脱落,直接生成了化合物8,简化操作的同时,大大提高了收率。
2.3 9的合成
在化合物9的合成过程中,文献[14]采用了1,4-二氮杂二环[2.2.2]辛烷(DABCO)和乙腈(MeCN)分别作为催化剂和溶剂,反应时间较长(5 h),且DABCO和MeCN均具有一定的毒性,DABCO在常温下易潮解易升华,实验安全性低,不便操作。参照文献[19]采用DMF作溶剂,将化合物8溶解,0 ℃下依次加入咪唑和TBDMSCl,明显缩短了反应时间(2 h),但收率仅为52%,且有较多副产物生成,因此,参照文献[12,20]对化合物9的合成工艺进行了探索见表1。
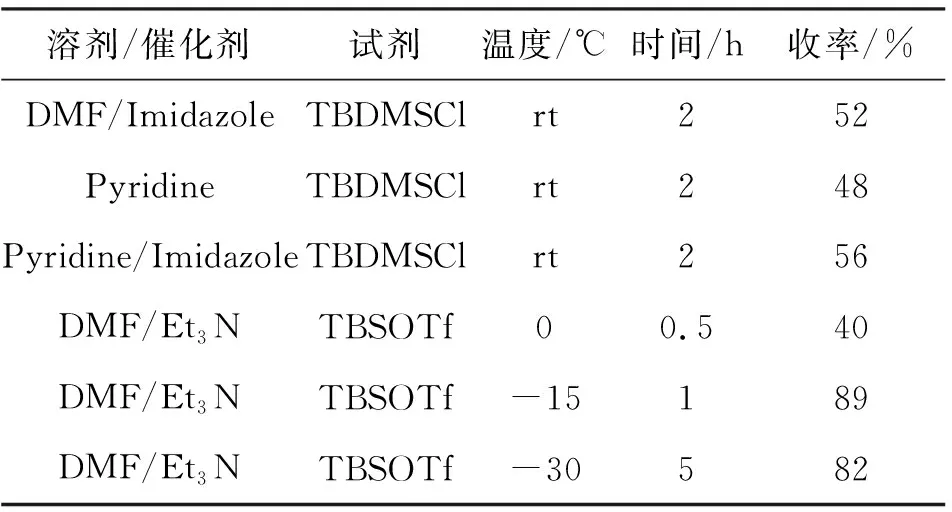
表1 合成化合物9的反应条件筛选Table 1 Reaction conditions to synthesis compounds 9
在尝试以上条件之后发现,当化合物8与TBDMSCl反应时,无论是以DMF还是吡啶作溶剂,均有较多原料剩余,且有副产物生成,从而导致产率降低。将TBDMSCl换成TBSOTf之后,在0 ℃下依然有较多副产物生成,但通过降低温度以及分批加入TBSOTf可以大大提高收率。因此,经过以上探索,最终确定,采用DMF作溶剂,干燥的Et3N作催化剂,在-15 ℃下分批加入TBSOTf,可以保证反应完全,产物单一,收率高且便于纯化。
以廉价易得的D-半乳糖为原料,通过10步反应,以总收率18.6%首次完成了目标化合物10的合成;高效地在2-位,5-位和6-位引入了临时保护基,实现了正交保护,且5-位的喹啉-2-甲酰基可以高立体选择性地控制1,2-cis半乳呋喃糖苷键的生成;对化合物5, 8, 9的反应条件进行了优化,极大提高了目标化合物的收率和实验安全性。本路线具有操作简便、原料易得、安全性高等优点,也为寡糖片段F1SS的后续合成奠定了基础。
