新型1,2,4-三取代-1,2,3-三氮唑盐的合成
2020-05-13敖慧龙袁克萌王飞军石晓濛
敖慧龙, 张 磊, 袁克萌, 王飞军, 石晓濛
(华东理工大学 化学与分子工程学院,上海 200237)
含氮杂环卡宾的催化反应在过去50年内得到了广泛的关注和长足的发展。近年来,Enders等的研究工作[1-4],又将这一领域推向了新的高度,噻唑卡宾、咪唑卡宾、1,2,4-三氮唑卡宾催化反应的出现,为广大科研工作者们提供了崭新的合成思路。
2011年,Bertrand[5-6]合成并分离了一种以1,2,3-三氮唑为基础结构的新型介离子卡宾,这种独特结构的卡宾又被称为远端含氮卡宾或异端含氮卡宾。这类新型卡宾中的杂原子分布在环内同一侧,并能以一种独特的共振结构稳定存在,也正因为这种特别的电子排布结构,使它们成为了一种理想的亲核有机催化剂。2013年,Robin[7]以合成的三氮唑卡宾盐为催化剂,有效的促进了烯醛与查尔酮的环加成反应。2014年,Jason[8]用合成的并环体系1,2,3-三氮唑卡宾前体盐催化醛

Scheme 1

Chart 1

Scheme 2
的氧化酯化反应,首次利用分子内环化的空间选择性,较高产率的合成了1,2,4-三取代的1,2,3-三氮唑卡宾前体盐(Chart 1)。
越来越多的研究结果表明,在催化反应过程中,自由状态的卡宾反应性活泼,化学寿命短,因此三氮唑卡宾盐作为催化剂的适应范围并不十分广泛,这一状况随着三氮唑卡宾金属络合物的出现才有所好转[9]。三氮唑卡宾在与一些过渡金属络合后,形成了一种更加稳定的结构,并使其具备了更出色的催化性能[10]。2013年,Martin等[11]以三氮唑卡宾铑络合物在无碱无氧化剂的条件下,催化氧化仲醇的反应,取得了显著效果。
1,2,3-三氮唑卡宾出现后,各种1,3,4-取代的1,2,3-三氮唑卡宾盐的研究层出不穷,但1,2,4-取代的1,2,3-三氮唑盐的设计合成与分离以及催化反应却鲜有人报道。本文以邻溴苄醇(1)和水杨醛(2)为原料,经5步反应合成了一种分子内同时含羟基和1,2,3-三氮唑环的新型类卡宾前体盐中间化合物(7),在三氟甲磺酸酐的催化下,分子内六元环化反应得到一种新型的1,2,4-三取代-1,2,3-三氮唑并三环体系类卡宾前体盐(8, Scheme 1),其结构经1H NMR,13C NMR和MS(ESI)表征。
1 实验部分
1.1 仪器与试剂
Bruker AM-400 MHz型核磁共振仪(CDCl3为溶剂,TMS为内标);Finnigan MA+型质谱仪。
2[12]、 3[13]、 4[14]、 5[15]、 6[16]、 7[17]按文献方法合成;其余所用试剂均为分析纯或化学纯。
1.2 合成
将3.14 mmol的化合物7和N,N-二异丙基乙胺0.20 mL(3.45 mmol)溶于15 mL无水乙腈中,氮气保护下,冰浴冷却15 min,于0 ℃滴加Tf2O 0.42 mL(3.45 mmol),滴毕,反应2 h(TLC检测)。旋干溶剂,残余物用DCM稀释,析出淡黄色晶体,抽滤,滤液旋蒸除溶得化合物8。
8a: 白色固体,收率65.3%;1H NMR(400 MHz, CDCl3)δ: 7.86(s, 1H), 7.79(d,J=8.2 Hz, 2H), 7.52(d,J=7.8 Hz, 1H), 7.29~7.42(m, 6H), 5.94(t,J=7.7 Hz, 1H), 4.64(dd,J=6.4 Hz, 14.4 Hz, 1H), 4.50(dd,J=5.6 Hz, 14.8 Hz, 1H), 2.48~2.57(m, 1H), 2.26~2.38(m, 1H), 0.99(t,J=7.1 Hz, 3H);13CNMR(100 MHz, CDCl3)δ: 147.8. 137.1, 136.3, 130.8, 130.4, 129.0, 128.8, 128.2, 126.6, 125.6, 118.4, 61.9, 41.3, 28.4, 23.2, 11.0; HR-MS(TOF)m/z: Calcd for C18H19N3{[M+H]+}277.1528, found 277.1534。
Scheme 3
8b: 淡黄色固体,收率59.4%;1H NMR(400 MHz, CDCl3)δ: 7.78(d,J=10.4 Hz, 2H), 7.66(s, 1H), 7.44(s, 1H), 7.23~7.41(m, 9H), 7.12(s, 2H), 6.94(d,J=10.2 Hz, 1H), 4.39(s, 2H);13C NMR(100 MHz, CDCl3)δ:147.5, 137.6, 136.5, 136.1, 130.6, 130.3, 129.1(d,J=7.7 Hz), 128.9(d,J=9.9 Hz), 128.5, 128.3, 128.2, 125.7, 120.5, 64.5, 41.2; HR-MS(TOF)m/z: Calcd for C22H19N3{[M+H]+}325.1529, found 325.1534。
2 结果与讨论
2.1 五元环化反应
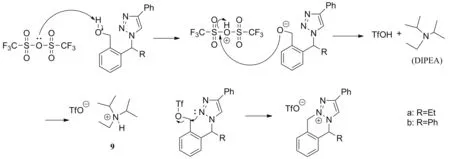
基于Scheme 1的合成路径,设计了以水杨醛为原料的合成反应,如Scheme 2所示。在该合成路径中,最终只能得到产物7c与7d,无法通过分子内闭环合成五元环的分子内盐8c与8d,因此判断从7a和7b到六元环分子内盐8a与8b的闭环反应中,机理如Scheme 3所示。基于该反应机理猜测7c与7d无法闭环的原因,可能是由于苯环的强共轭效应使得与三氟甲磺酸基团相连的苯环碳原子不具备足够的电负性,无法接受N2上的孤对电子,不能使三氟甲磺酸基团离去闭合成环。
2.2 闭环反应中溶剂的选择
在闭环反应中,同等实验条件下不同溶剂对于反应的产率有较大影响,在可用溶剂范围内,随着溶剂极性的增加,反应产率逐渐上升,这可能与反应物和产物在溶剂中的溶解度有关,结果如表1所示。
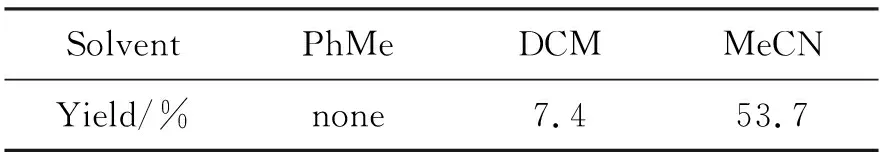
表1 溶剂对8a产率的影响Table 1 The yield of 8a in different solvents
Scheme 4

Scheme 5
2.3 缩减反应步骤的尝试
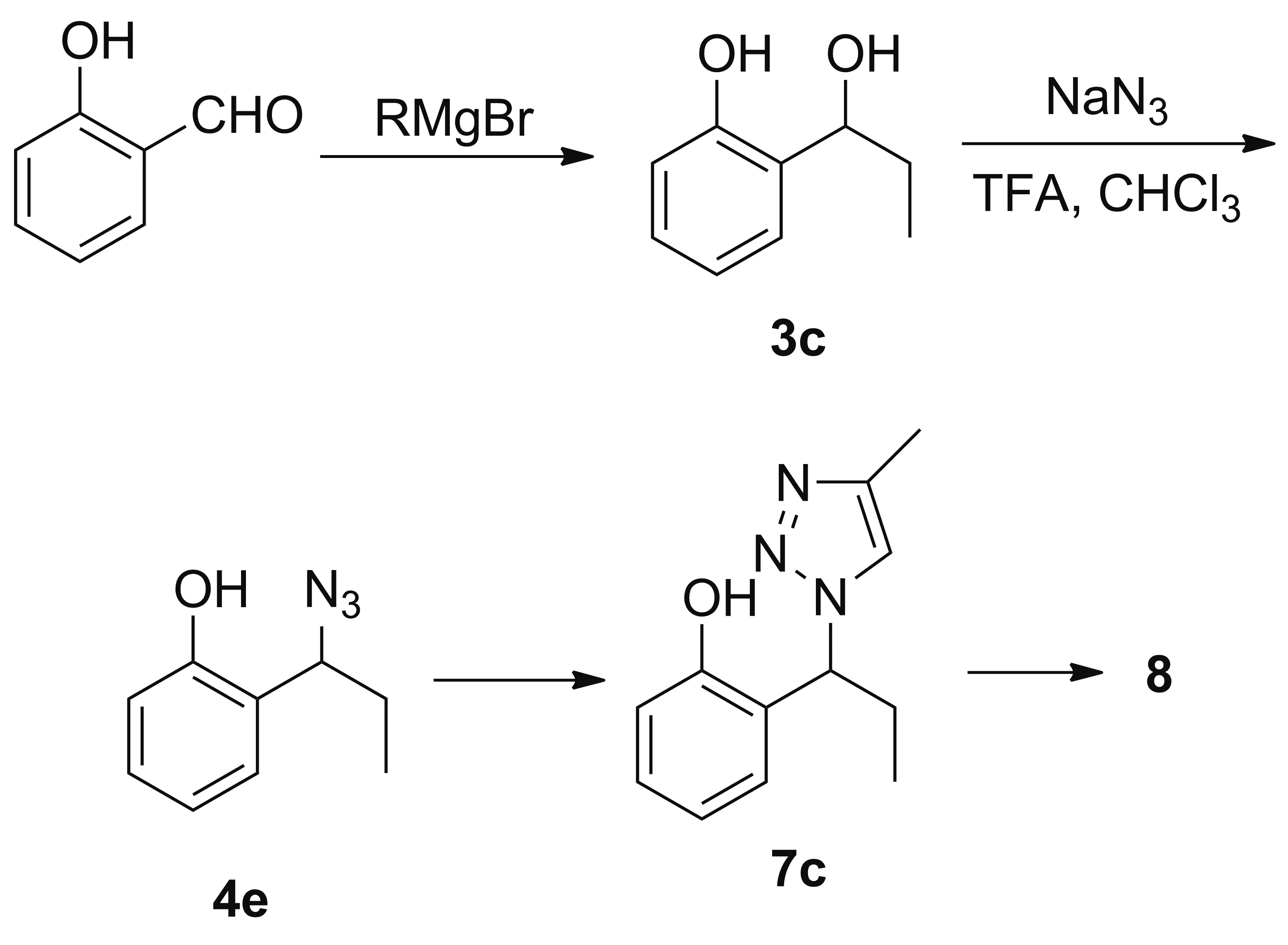
考虑到酚羟基和苄位羟基不同的化学性质,以及Scheme 1中氯甲基甲醚(MOM)对苄位羟基的保护,参考文献[18]合成酚类叠氮化合物的方法,对Scheme 2的合成路线进行了缩减尝试(Scheme 4)。该合成路线成功分离得到了酚类叠氮化合物4e,由4e经Huisgen反应却不能得到目标产物7c。由此推测,一是酚羟基的酸性太强,在逐滴加入4e的甲苯溶液后,酚羟基干扰了苯乙炔与CuTc的后续反应过程,从而让环加成反应难以为继;二是酸性的酚羟基与炔铜发生亲核加成反应,大大的阻碍了叠氮基团与炔铜的环加成反应,使得Huisgen反应无法正常进行。
Scheme 6
2.4 氯甲基甲醚在两种路线中的不同作用
通过缩减反应步骤的尝试可以断定氯甲基甲醚(MOM)在Scheme 2中起到的并不是保护酚羟基的作用,而是在成醚反应后有效的抑制酚羟基的酸性,令Huisgen反应得以顺利的进行下去。与之相对的,在Scheme 1中,苄位羟基上的氢远不如酚羟基氢活泼,对于后续的Huisgen反应也并无影响[19],所以氯甲基甲醚(MOM)是在生成叠氮类化合物5a, 5b之前的反应过程中,起到保护苯环上苄位羟基的作用。
2.5 新型三氮唑盐催化反应活性的探索
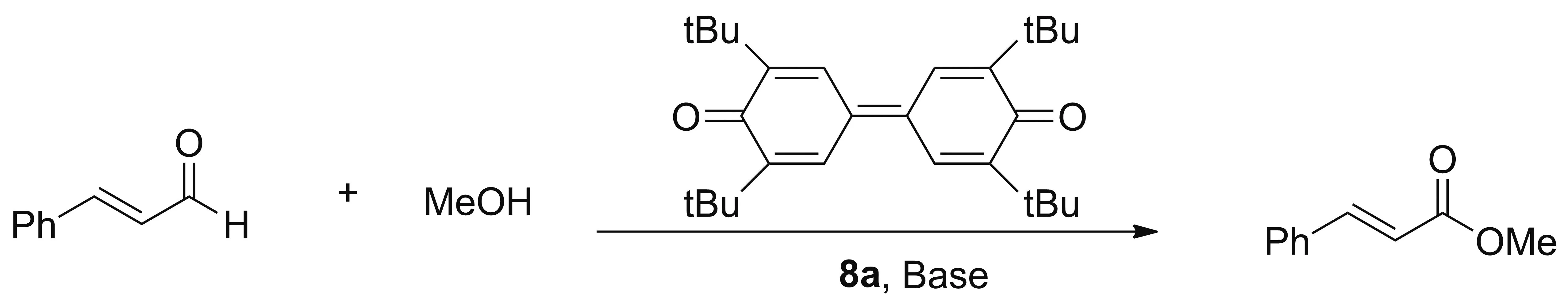
针对三氮唑卡宾前体盐8参考文献[8,20]设计了其与金属的络合反应(Scheme 5)以及作为小分子催化剂催化α,β-不饱和醛与甲醇的酯化反应(Scheme 6)。对实验条件进行筛选,发现两个反应均未得到理想中的目标产物,在确定操作和实验条件都准确无误的情况下,我们猜测造成反应无法进行的原因可能有以下两点:(1)三氮唑盐8中六元环上两个与苯环相连的α-C和三氮唑环产生超共轭效应,三氮唑环上的电子云密度增加,降低了碳碳双键上氢的酸性,使反应受阻;(2)如Scheme 7所示:在碱性条件下,1,3,4-取代的1,2,3-三氮唑盐(10),可以通过环内电子转移,得到带有卡宾碳的中间体11,因此能与金属发生有效的络合反应并具有相应的催化活性。而1,2,4-取代的1,2,3-三氮唑盐(8),因为取代基位置的原因,N2位带正电荷,导致三氮环的电子云密度更加对称,环上的偶极矩减小,对环内电子的转移有一定的阻碍作用,使得双键碳上氢的酸性降低,所以即便增加反应体系的碱性,氢离子也难以被夺去,无法生成带有碳卡宾结构的中间体12,导致其与金属的络合能力与催化活性均大幅度降低。

Scheme 7
设计了一条合理的合成路线,以邻溴苄醇为基础原料合成了一种并三环体系的新型1,2,4-三取代-1,2,3-三氮唑卡宾前体盐,在此基础上对其催化反应活性进行了初步的探索与研究。
