特发性偏身肥大症合并脑白质病变1例报告
2020-05-12王晓玉郭雯琪王迎新
王晓玉, 郭雯琪, 曹 华, 王迎新
特发性偏身肥大症(idiopathic hemihypertrophy,IH)是一种先天罕见疾病,以一侧肢体、躯干和头面部或者整个半身不对称生长、肥大为主要特征,肥大包括软组织以及骨骼[1,2]。偏身肥大症(hemihypertrophy,HH)可以独立存在,也可以是某些先天性异常综合征的一部分,前者称为IH[1,2]。该病发病率极低,为1/13000~1/86000[3,4],临床医生对此疾病认识少,患者就医困难。现报道1例IH合并脑白质病变患者。
1 临床资料
患者,男,27岁。因“发现右下肢增粗17 y” 于2019年5月21日入住大连医科大学附属第一医院神经内科。患者17 y前10岁时发现双下肢长度不等,右下肢较左下肢长约1 cm,此后发现右下肢直径较左下肢增粗,15岁到18岁期间右下肢增粗明显。多次于外院就医,未明确诊断。无肢体疼痛、麻木、无力、二便障碍。无抽搐、无意识丧失。既往史:体健。个人史:为父母第2胎,孕期正常,母亲无放射线及毒物接触史。足月顺产,出生时体重3.2 kg,1岁半会走路。幼时经常跌倒,运动能力较同龄人略差。智力正常,理工科大学本科毕业,工程师。未婚。家族史:父母非近亲结婚,有一姐姐体健。家族中无同类患者。查体:颈、胸、背部多处可见皮下结节,质软,不可移动。胸部剑突水平皮肤见直径5 cm类圆形皮肤色素沉着。双手掌大小鱼际肌较平,掌纹深,双肘屈曲侧皮纹少、浅。右下肢较左下肢长2 cm(R:105 cm;L:103 cm);右下肢周径较左下肢明显增粗:双下肢膝以上10 cm周径:右侧较左侧圆周长11.5 cm(R:57.5 cm;L:46 cm);双下肢膝部周径:右侧较左侧圆周长16.5 cm(R:54.5 cm;L:38 cm);双下肢膝以下10 cm周径:右侧较左侧圆周长8 cm(R:47 cm;L:39 cm);脚踝周径:右侧较左侧长5.5 cm(R:30 cm;L:24.5 cm)。右膝关节表面皮纹浅,双足轻度马蹄内翻足,右足皮肤粗糙、脱屑、体毛重(见图1)。神经系统查体:神清,语明。精神、智能正常。颅神经查体未见明显异常。右侧L2~S2水平痛觉减退。双上肢及右下肢腱反射减弱。右侧Babinski征(+),左侧Babinski征(-),双侧Chaddock征(+)。其余神经系统查体未见明显异常。辅助检查:头部MRI+MRA:双侧额顶叶皮质及皮质下异常信号,T2Flair斑片状高信号(见图2);头部MRA未见异常。颈椎MRI:颈5/6椎间盘轻度膨出;胸椎MRI未见异常。腰椎MRI:腰5骶1椎间盘突出。 双下肢股骨、胫腓骨MRI:右侧大腿肌肉脂肪化,右侧小腿前内侧皮下脂肪增厚。右侧股骨、胫骨直径较左侧增粗(见图1)。肌电图:右下肢未见明显神经源性或肌源性损害。脑电图:界限性脑电地形图,界限性脑电图,背景快波活动增多。心电图:正常。胸部CT(平扫+HR):双肺多发磨玻璃结节,双肺下叶少许炎症改变。腹部彩超、泌尿系彩超未见异常。心脏彩超、颈部大血管彩超、双下肢动脉、深静脉彩超未见明显异常。化验检查:血、尿、便常规;肝、肾功能;血糖、血脂、电解质、肌酸激酶、心肌标志物、血沉;肝炎病毒系列、梅毒、艾滋病毒;风湿、免疫、肿瘤标志物均未见明显异常。颈部皮肤结节切除行皮肤病理染色:表皮囊肿伴肉芽肿性炎症。遗传病全外显子组基因测序(金域医学):检测到一个杂合基因变异:9q34 TSC1 Intron10 c.1029+3 A>G,为内含子区微小点突变,临床意义未明。其父亲、母亲均未检测到该杂合基因突变。
2 讨 论
IH是一种先天罕见疾病,以一侧肢体、躯干和头面部或者整个半身不对称生长、肥大为主要特征,肥大包括软组织以及骨骼[1,2]。其潜在的病理机制是肥大组织细胞增殖速率增高,而不是细胞体积增大[3,4]。HH可以独立存在,称为IH,也可以是某些先天性异常综合征的一部分[1~7]。该病发病率极低,大约为1/13000~1/86000[3,4]。多毛症(hypertrichosis)是指在与同种族、同性别、同年龄个体相比毛发过度生长,多毛症可以分布于全身,也可以局限于一小片,目前认为先天性多毛症是一种遗传性疾病或是散发性基因突变的结果。迄今为止,全世界仅有4例IH合并偏身多毛症(hemihypertrichosis)的病例报告[2]。因此,临床医生对此疾病认识少,本例患者在就诊于我科之前多次辗转于各医院门诊都没有得到确诊,直到27岁才得以明确诊断,这其实面临着极大的疾病风险,因为HH患者在儿童与少年时代易患儿童性恶性肿瘤(如Wilms肿瘤、肝母细胞癌、神经母细胞瘤和肾上腺癌等[1~7]),需要定期进行肿瘤监测。此例患者入院后经过系统检查未发现恶性肿瘤,仅合并肥大侧肢体体毛重,因此诊断为特发性偏身肥大症合并偏身多毛症(idiopathic hemihypertrophy associated with hemihypertrichosis)。另外,此前报告的4例患者3位为婴幼儿时期就诊,1例16岁时就诊,4位患者就诊时头部MRI检查无阳性发现,本例27岁男患虽无中枢神经系统症状,但在查体时发现有双侧锥体束受损体征,头部MRI T2Flair显示双侧额顶叶皮质及皮质下斑片状高信号病灶。该患者查体发现的神经系统受累体征可以以额顶叶皮质及白质病变解释。该患者颈、胸、背部多处可见的皮下结节经取颈部皮下结节活检为表皮囊肿伴肉芽肿性炎症。
包含偏身肥大症状的常见先天综合征包括:Beckwith-Wiedemann综合征(Beckwith-Wiedemann Syndrome,BWS)、Klippel-Trenaunay-Weber综合征以及Proteus综合征等[1~7]。BWS以偏身肥大、内脏肥大、巨大畸形新生儿、巨舌、脐疝和胚胎来源的肿瘤为主要表现[1,2,4],80%以上的患者发现染色体11p15.5分子异常,即父系来源的单亲二体性(paternal uniparental disomy,pUPD 11p15)[4]。Klippel-Trenaunay-Weber综合征以肢体或手足软组织肥大、葡萄酒色痣和静脉曲张畸形为主要临床表现,与VG5Q基因激活物突变有关[1,2]。Proteus综合征表现为长骨肥大,局部尤其是手足巨大畸形、足底畸形生长,血管瘤、脂肪瘤、线状疣状表皮痣和局部皮肤发育不全,相关致病基因未明[1,4]。与HH相关的基因缺陷由Martin等[8]于2005年首先报告。Bliek等对73例HH患者进行了表观遗传学分析,15%的HH患者检出pUPD 11p15缺陷;6%患者检出KGNQ1OT1去甲基化;约3%患者检出H19过甲基化[5]。
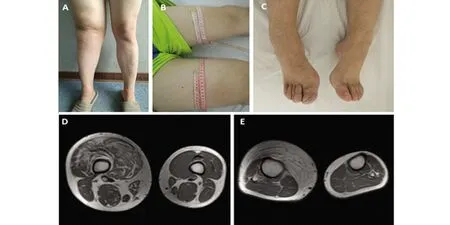
A:右下肢较左下肢明显增粗;B:膝以上10 cm周径:右侧57.5 cm,左侧46 cm;C:右足较左足大,右足背体毛重于左足;D:双侧股部MRI示右股部周径大于左股部,右股骨直径大于左股骨,右股部肌肉脂肪化;E:双小腿MRI示右小腿周径大于左小腿,右胫骨直径大于左胫骨,右小腿前部皮下脂肪明显增生
图1 患者双下肢外观、双股部与双小腿MRI横断面
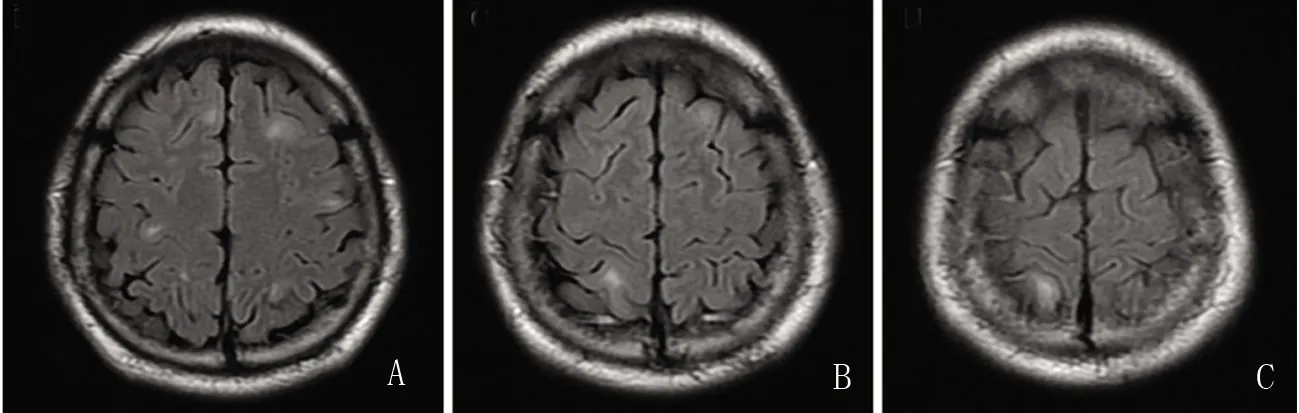
图2 患者头部MRI T2 Flair序列:A、B、C显示双侧额叶、顶叶皮质及皮质下白质散在高信号改变
HH被认为是由马赛克式(a mosaic form)的基因缺陷导致的马赛克式的过度生长,实际上,研究发现pUPD总是以马赛克形式存在,增大的组织器官中由较大比例的二体性细胞(disomic cells)组成[5]。Itoh 等[9]研究1例BWS不同组织甲基化形式,他们展示了肥大器官含有pUPD的细胞比例较高(40%~50%),pUPD也以较低百分比存在于血液淋巴细胞中,可以通过常规检测做出诊断。当pUPD仅以很低的比例存在于细胞中,很可能不存在于血液淋巴细胞中。Shuman等[10]设想对于一部分不能解释的IH病例pUPD仅存在于肥大组织中,因此,在常规的血液诊断性检测中不能被检测到。
该患者外周血基因检测未查到pUPD 11p15。所查到的9q34 TSC1 Intron10 c.1029+3 A>G,为内含子区微小点突变,临床意义未明。其父亲、母亲均未检测到该杂合基因突变。患者右下肢肥大不影响生活与工作,目前无需特殊治疗,其神经系统体征及头部MRI病变我们将长期随访。
