柱前衍生化-超高效液相色谱-三重四极杆串联质谱法测定人血清中11种雌激素的方法学研究*
2020-04-29杨娜王敏王月园柳航朱怀军葛卫红
杨娜,王敏,王月园,柳航,朱怀军,葛卫红
(南京大学医学院附属鼓楼医院药学部,南京 210008)
雌激素作为人体内重要的性激素之一,具有重要而广泛的生理作用。研究表明,雌激素不仅有维持女性生殖功能和第二性征的生理作用,而且对代谢系统的调节、神经系统的发育、骨骼的生长等有一定程度的影响[1-2]。雌二醇(Estradiol,E2)是体内活性最强的雌激素,其通过细胞色素P450酶、甲基转移酶等重要酶类进一步发生代谢[3]。目前。越来越多的研究证实,除雌二醇外,多种雌激素代谢产物同样具有生物学活性,且与相关疾病的发生发展密切相关。例如2-羟基雌酮(2-Hydroxyestrone,2-OHE1)和16α-羟基雌酮(16α-Hydroxyestrone,16α-OHE1)比例降低可能增高乳腺癌的发病风险[4], 4-甲氧基雌酮(4-Methoxyestrone,4MeOE1)可参与脂质的调节等[5]。因此,对血清中雌激素代谢网络各类代谢物的浓度分析和整体代谢轮廓描绘,对辅助临床诊断、发现疾病生物标志物以及疾病发病风险的评估具有重要意义。临床上内源性类固醇激素的测定技术,包括液相紫外检测法、气相色谱法以及免疫测定法等[6-8]。其中,免疫测定法最常用于类固醇的检测,但由于易受相关内源性类固醇、脂质和基质效应的干扰,其对于类固醇激素测定的特异性和可靠性仍然存在疑问。虽然部分研究采用HPLC-MS/MS法测定体内激素含量,但往往存在测定种类少、分析时间长、灵敏度低等问题。本研究旨在建立快速、高灵敏度的血清雌激素UPLC-MS/MS分析方法,并对此方法进行方法学考证,为雌激素的临床检测及雌激素相关疾病的研究提供支持。
1 材料与方法
1.1实验材料
1.1.1实验试剂 雌二醇(Estradiol,E2,纯度:96.3%,批号:100182-201205,中国食品药品检定研究院);雌酮(Estrone,E1,批号:100849-200501,中国食品药品检定研究院);雌三醇(Estriol, E3,纯度:97.6%,批号:100934-201302,中国食品药品检定研究院);2-羟基雌酮(2-Hydroxyestrone,2-OHE1,纯度:98%,批号:3-SBT-43-2,加拿大TRC);2-甲氧基雌酮(2-Methoxyestrone,2MeOE1,纯度:98%,批号:2-AKS-176-1,加拿大TRC);4-甲氧基雌酮(4-Methoxyestrone,4MeOE1,纯度:97.72%,批号:1-SRE-175-1,加拿大TRC);16-表雌三醇(16-Epiestriol,16epiE3,纯度:96%,批号:29-AZC-158-2,加拿大TRC);17-环雌三醇(17-Epiestriol,17epiE3,纯度:95%,批号:20-THT-39-2,加拿大TRC);2-羟基雌二醇(2-Hydroxyestradiol,2-OHE2,纯度:96%,批号:6-SBT-57-6,加拿大TRC);16α-羟基雌酮(16α-Hydroxyestrone, 16α-OHE1,纯度:97%,批号:3-CGF-67-6,加拿大TRC);2,3,4-13C3-雌酮(2,3,4-13C3-Estrone,E1-13C,IS,纯度:98%,批号:MBBC4138,美国CIL);丹磺酰氯(批号:BCBL7618V,Sigma);叔丁基甲醚(阿拉丁);Waters Oasis HLB固相萃取小柱(美国Waters);葡聚糖-活性炭(批号:SLBV2046, Sigma);色谱柱C18(Waters ACQUITY UPLC BEH C18);L-抗坏血酸(批号SLBM0850V, Sigma);甲醇、乙腈(色谱纯,德国Merck)。
1.1.2主要仪器 Waters ACQUITY UPLC I-Class /XEVO TQD超高效液相质谱联用仪(Waters,美国),CV200真空浓缩仪(北京吉艾姆科技有限公司),TGL-16.5M冷冻离心机(上海卢湘仪离心机仪器有限公司),DW-HL340超低温冰箱(Thermo Fisher,美国)。
1.2实验方法
1.2.1色谱条件 色谱柱:Waters BEH C18columns(2.1 mm×50 mm,1.7 μm);柱温:40 ℃;流动相:乙腈-水(含0.1%甲酸),采用70%(乙腈)等度洗脱,流速0.3 mL·min-1;整个运行时间:5 min;进样体积5 μL。
1.2.2质谱条件 离子源:点喷雾离子源(ESI);扫描模式:正离子模式;检测方式:多重反应监测(MRM);毛细管电压为3.2 kV;脱溶剂气为氮气,脱溶剂温度为350 ℃,流速为800 L·h-1;碰撞气为氩气。各种雌激素的相关离子如表1所示。

表1 MRM模式离子参数
1.2.3储备液和工作液配制 分别精密称取雌激素标准品和同位素标记的内标E1-13C(IS)粉末2 mg,置于20 mL量瓶中,加入含0.1%L-抗坏血酸的甲醇溶液定容至刻度线,颠倒摇匀配成终浓度为100 μg·mL-1的储备液,-20 ℃储存。分别取各类雌激素储备液,依次用L-抗坏血酸甲醇梯度稀释,分别配成浓度为0.2,0.5,1,2,5,10,20 ng·mL-1的雌激素混合标准曲线工作液。取内标储备液,采用含0.1%L-抗坏血酸的甲醇溶液逐级稀释至10 ng·mL-1的内标工作液。衍生化试剂配制:精密称取丹磺酰氯试剂10 mg于15 mLEP管中,加入丙酮溶解,配置成1 g·L-1的丹磺酰氯衍生化试剂溶液,置于-20 ℃储存。
1.2.4空白血清的制备 取空白血清10 mL,加入0.1 g 葡聚糖-活性炭后,旋涡混匀,于37 ℃摇床5 h后4000 r·min-1离心15 min,上清液于12 000 r·min-1转速下低温超速离心30 min,取上层澄清液,即为空白血。
1.2.5血清样品的处理方法 取300 μL血清样本,加入20 μL内标(10 ng·mL-1)混匀后,加入甲基叔丁基醚溶液1.5 mL涡旋振荡3 min,于14 000 r·min-1,4 ℃离心10 min,取上清液1.3 mL于真空浓缩装置中挥干。依次加入50 μL 0.1 mol·L-1的碳酸氢钠缓冲液pH值9.0和50 μL丹磺酰氯衍生化试剂后振荡,于60 ℃水浴加热10 min衍生化。取出冰浴,并加入50 μL含0.1%甲酸乙腈-水溶液(1:1),涡旋混匀后,于14 000 r·min-1,4 ℃离心5 min,取上清液5 μL进样分析。
2 结果
2.1分析方法优化
2.1.1质谱条件优化 采用浓度为500 ng·mL-1的标准溶液,进行衍生化后,对11种雌激素及内标的母离子、子离子、锥孔电压和碰撞能量进行逐一优化和确认,使目标化合物具有最高灵敏度,见表1。各类雌激素在衍生化处理后,极大提高了离子化效率,且进入质谱均能产生相应强度高且稳定性好的[M+H]+分子离子峰,衍生化试剂与母核结合进入二级质谱碎裂为171子离子碎片。
2.1.2液相条件优化 本实验选用Waters ACQUITY UPLC BEH C18色谱柱,分别使用甲醇和乙腈作为有机相对梯度和等度不同流动相体系进行比较研究。结果发现,同时在乙腈和水中加入0.1%甲酸,并在70%乙腈等度洗脱时,能够很好的分离各类激素,且各物质峰形良好具有较高灵敏度。本方法在UPLC液相系统下,分析时间可以缩短到5 min,能够快速高效的分析各雌激素,达到很好的定量分析效果。
2.1.3样品处理优化 采用葡聚糖-活性炭法能够高效去除人血清中内源性雌激素的干扰,比BSA代替血清更加合理[9]。本实验前期分别使用蛋白沉淀法、固相萃取法(Waters HLB 固相萃取柱)、二氯甲烷和叔丁基甲醚对血清样本前处理进行摸索。蛋白沉淀法杂质干扰多,基质效应较高。液-液萃取法的重现性和精密度均优于固相萃取,其中叔丁基甲醚萃取法具有较好的提取回收率,使各类雌激素的提取回收率均大于70%,并能保证较低的基质干扰。此外,本实验通过对衍生化的温度、时间、试剂浓度等进行优化得到最优的衍生化条件。
2.2方法学验证
2.2.1特异性 按“1.2.5”项处理空白血清样本和加入各类雌激素标准品血清样本,如图1所示,E1、E2、E3、16epiE3、17epiE3、2MeOE1、4MeOE1、16α-OHE1、2-OHE1、4-OHE1、2-OHE2和IS的保留时间分别为2.78,2.36,1.07,1.42,1.52,2.55,2.77,1.40,2.16,2.35,2.36 min和2.78 min。待测物和内标峰形良好,空白血清在各雌激素离子通道下无干扰,各类雌激素在该方法下特异性良好。
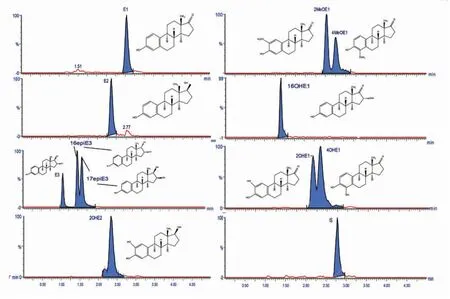
图1 UPLC-MS/MS测定11种雌激素及内标的典型色谱图
Fig.1Typicalchromatogramsof11estrogensandISinUPLC-MS/MSdetermination
2.2.2标准曲线和定量下限 取空白血清270 μL,分别加入含雌激素混合标准溶液30 μL,使血清中浓度分别为20,50,100,200,500,1000,2000 pg·mL-1,按照“1.2.5”项下处理样本以检测目标峰面积和内标峰面积的比值为纵坐标(Y),血清浓度为横坐标(X),进行线性回归(权重1/X)。得到各类雌激素的标准方程如表2,各种雌激素在20~2000 pg·mL-1范围内具有良好的线性,定量下限(LLOQ)均为20 pg·mL-1(S/N>10)。
2.2.3精密度和准确度 取空白血清于3 d分别配制3批含LLOQ、低、中、高浓度(20,50,200,1 600 pg·mL-1)的混合雌激素的血清样品,每个浓度平行6份,按照“1.2.5 ”项下处理样本,测定后计算精密度与准确度。由结果可知,精密度与准确度均符合生物样本测定需求。见表3。

表2 标准曲线方程
2.2.4提取回收率和基质效应 分别按照以下方法配制处理雌激素样本,并计算提取回收率和基质效应,计算结果见表4。
A:取空白血清,按“2.2.2”项下标准曲线处理方法,配制含有终浓度为50,200,1 600 pg·mL-1的混合雌激素血清样本各6份,加入20 μL内标(10 ng·mL-1)混匀后按照“1.2.5”项处理血清样品。取上清液5 μL进样分析,记录各类雌激素的峰面积A1和内标峰面积AIS1。
B:取空白血清,加入1.5 mL甲基叔丁基醚溶液涡旋振荡3 min,于14 000 r·min-1,4 ℃离心10 min,取上清液1.3 mL,分别加入雌激素混合标准工作液30 μL(浓度为0.5,2,16 ng·mL-1)、内标工作液20 μL混合后于真空浓缩装置中挥干。依次按“1.2.5”项加入50 μL,0.1 mol·L-1的碳酸氢钠缓冲液(pH值9.0)和50 μL丹磺酰氯进行衍生化反应及后续处理,取上清液5 μL进样分析,记录各类雌激素的峰面积A2和内标峰面积AIS2。
C:向离心管中直接加入雌激素混合标准工作液30 μL(浓度为0.5,2,16 ng·mL-1)、内标工作液20 μL,真空浓缩挥干。残渣依次按“1.2.5”项加入50 μL 0.1 mol·L-1的碳酸氢钠缓冲液(pH值9.0)和50 μL丹磺酰氯进行衍生化反应及后续处理,取上清液5 μL进样分析,记录各类雌激素的峰面积A3和内标峰面积AIS3。
表3 精密度和准确度

Tab.3 Accuracy and precision n=6
待测物的提取回收率=A1/A2(mean)×1.3/1.5 ×100%(1.3/1.5为萃取率)
待测物的基质效应=A2/A3(mean)×100%
内标的提取回收率=AIS1/AIS2(mean)×1.3/1.5 ×100%(1.3/1.5为萃取率)
内标的基质效应=AIS2/AIS3(mean)×100%
计算结果显示,采用叔丁基甲醚萃取法,11种雌激素的提取回收率均达到70%以上,并能保证较低的基质干扰。
2.2.5稳定性 配制含雌激素低、中、高浓度(50,200,1 600 pg·mL-1)的血清样本,分别置于室温24 h、反复冻融3次、-70 ℃冷冻保存1个月,4 ℃自动进样盘放置24 h,每种处理方式每个浓度平行6份。比较测定样本的精密度和准确度。结果显示低、中、高浓度的精密度(RSD)和准确度(RE)均在±15%内,如表5所示。
3 讨论
随着研究的深入,各类雌激素的生物学活性和生理学意义被不断地阐明。雌激素被认为和神经系统[10-11]、心血管系统[12]、骨代谢发生等关系密切[13]。在女性围绝经期,雌激素水平显著降低从而引起围绝经综合征。国内外指南推荐激素补充治疗是缓解绝经症状最有效的方法[14-15]。然而不断有研究表明,激素补充治疗会显著增加女性患乳腺癌、卵巢癌等疾病的风险[16]。因此,体内雌激素水平的评价和代谢轮廓的分析对激素补充治疗的个体化策略的制定具有重要意义。
自20世纪60年代以来,免疫法被广泛运用于临床类固醇激素的检测中,但其测定种类少、特异性低,且重要的是由于商品化试剂盒的来源、测定方法的差异导致跨实验室测定结果存在显著差异[17],因此国内尚无统一的性激素测定参考值。免疫法对于临床多中心、跨实验室的雌激素相关研究的开展,具有很大局限性。
随着质谱技术的发展,LC-MS/MS被逐渐运用到类固醇激素多组分分析中,从而使激素测定的种类显著增多,且特异性和准确性大大提高。对雌激素的研究,已经逐渐从传统关注的E2逐渐转向羟化代谢、甲基化代谢等重要代谢途径与疾病关联性分析中[18-19]。然而,很多有意义的临床研究所针对的雌激素测定种类少、灵敏度低或分析方法长,不能够全面而准确的了解雌激素代谢的整体轮廓[20-23],故其在相关疾病的危险因素分析或相关生物标志物的发现中不能提供很好的数据支持。本研究采用了UPLC-MS/MS结合柱前衍生化技术成功建立了可同时检测人血清中包括E1、E2和E3在内的11种雌激素的定量分析方法。该方法测定种类多、特异性强、精密度高,且分析时间仅5 min,显著优于传统免疫法。基于该方法的雌激素代谢整体轮廓的定量描绘在相关疾病的发病风险分析和激素补充治疗的个体化策略制定中具有一定的应用价值。
表4 提取回收率和基质效应
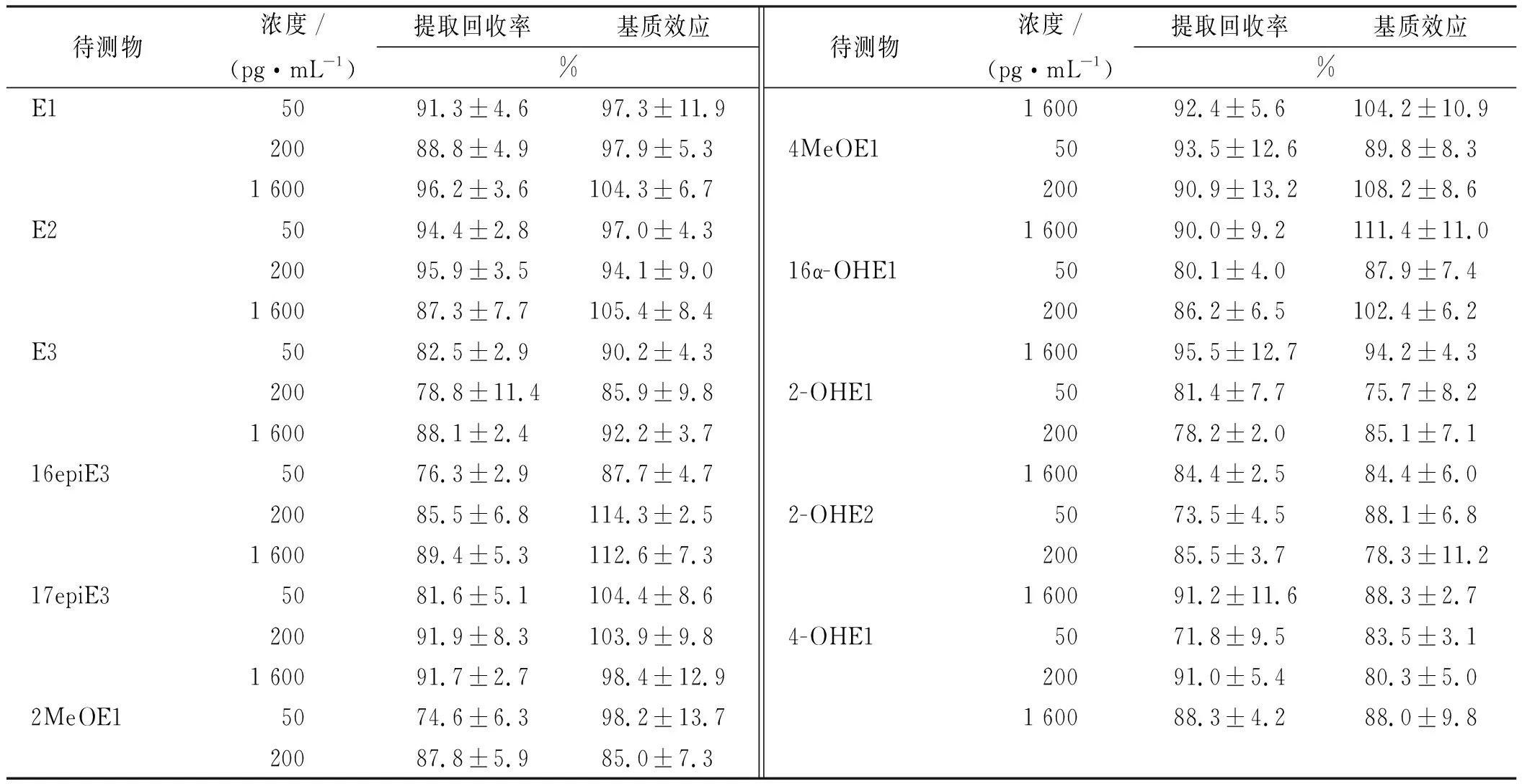
待测物浓度/(pg·mL-1)提取回收率基质效应%E15091.3±4.697.3±11.920088.8±4.997.9±5.31 60096.2±3.6104.3±6.7E25094.4±2.897.0±4.320095.9±3.594.1±9.01 60087.3±7.7105.4±8.4E35082.5±2.990.2±4.320078.8±11.485.9±9.81 60088.1±2.492.2±3.716epiE35076.3±2.987.7±4.720085.5±6.8114.3±2.51 60089.4±5.3112.6±7.317epiE35081.6±5.1104.4±8.620091.9±8.3103.9±9.81 60091.7±2.798.4±12.92MeOE15074.6±6.398.2±13.720087.8±5.985.0±7.3待测物浓度/(pg·mL-1)提取回收率基质效应%1 60092.4±5.6104.2±10.94MeOE15093.5±12.689.8±8.320090.9±13.2108.2±8.61 60090.0±9.2111.4±11.016α-OHE15080.1±4.087.9±7.420086.2±6.5102.4±6.21 60095.5±12.794.2±4.32-OHE15081.4±7.775.7±8.220078.2±2.085.1±7.11 60084.4±2.584.4±6.02-OHE25073.5±4.588.1±6.820085.5±3.778.3±11.21 60091.2±11.688.3±2.74-OHE15071.8±9.583.5±3.120091.0±5.480.3±5.01 60088.3±4.288.0±9.8
表5 雌激素稳定性实验结果
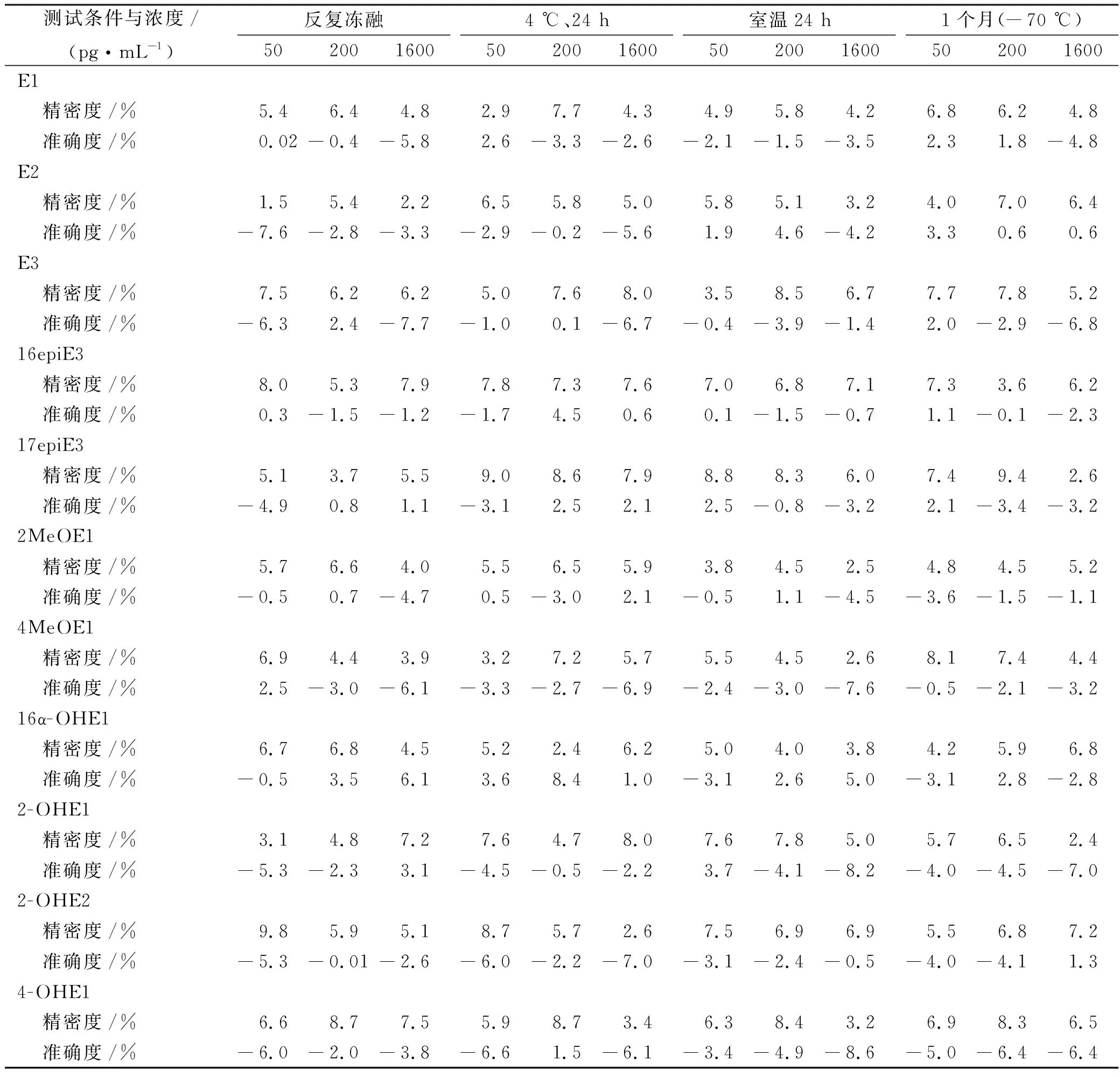
Tab.5 Stability results of the estrogens n=6
