线粒体功能障碍与糖尿病血管并发症关系研究进展
2020-04-28白春昀翁稚颖肖创贺源刘伟军郑昌博陈晨杨为民
白春昀,翁稚颖,肖创,贺源,刘伟军,郑昌博,陈晨,杨为民
昆明医科大学药学院暨云南省天然药物药理重点实验室,昆明 650500
2017年国际糖尿病联盟发布的第八版全球糖尿病地图显示,目前全球有4.25亿糖尿病患者,预计到2045年将会有近7亿患者[1]。糖尿病的血管并发症主要包括伤口愈合缓慢、糖尿病肾病、糖尿病心肌病、糖尿病视网膜病变、糖尿病脑病及糖尿病卒中等[2],超过一半的糖尿病患者死亡或致残为血管并发症所致[3]。内皮功能障碍是糖尿病血管并发症防治的重要环节[4],而线粒体功能障碍在糖尿病血管并发症中发挥重要作用[5-6],但目前对糖尿病及相关血管并发症尚无较好的治疗方案,因此,研究线粒体功能障碍如何影响糖尿病患者血管内皮功能,对糖尿病及其血管并发症的防治和药物研发具有重要意义。本文主要综述了线粒体功能障碍与糖尿病血管并发症中的关系。
1 线粒体的结构和生理功能
线粒体是由脂质双分子层膜结构组成的封闭结构,主要定位于大多数真核细胞的细胞质中[7]。线粒体从外到内可分为外膜、膜间隙、内膜及基质。外膜是线粒体最外层的膜,表面光滑,小孔富集,主要功能是维持线粒体膜间隙,也是物质进出、交换的通道。膜间隙主要含有细胞色素C等可溶性酶及半胱氨酸蛋白酶(caspase)-3、caspase-9的前体以及辅助因子,其中的腺苷酸激酶可催化三磷酸腺苷(adenosine triphosphate,ATP)分子末端磷酸基团转移到一磷酸腺苷(adenosine monophosphate,AMP),生成二磷酸腺苷(adenosine diphosphate,ADP)。内膜的主要功能是维持线粒体形态、动力学(分裂-融合的平衡)及形成ATP[8]。基质含有丰富的可溶性蛋白胶状物,有三羧酸循环相关酶,含DNA、RNA、核糖体等。
在生理状态下,线粒体功能主要有3个方面:①合成ATP。参与能量代谢的营养物质进入三羧酸循环后裂解成丙酮酸、脂肪酸进入线粒体,在基质内可由脂肪酸β氧化成还原型辅酶Ⅰ(nicotinamide adenine dinucleotide,NADH)及黄素腺嘌呤二核苷酸递氢体(flavin adenine dinucleotide,FADH2);裂解成辅酶A(CoA),进入三羧酸循环,也能产生NADH、H+或FADH2;以上反应能为电子传递链提供电子,产生跨膜质子梯度。质子通过线粒体ATP合酶回流,通过氧化呼吸链逐级传递,最终生成H2O2和CO2等终末产物,伴有能量的释放,可使ADP氧化磷酸化生成ATP。②调控Ca2+的储存及释放。线粒体还可通过内膜电化学梯度调节Ca2+的摄取、储存及释放以维持细胞内Ca2+浓度,同时Ca2+可调节线粒体内多种酶,其浓度会影响线粒体能量代谢[9]。③其他功能。线粒体通过Bax、caspase家族活化介导线粒体凋亡、线粒体自噬等多种生理病理过程,并参与细胞活动信号的转导。
在病理条件下,线粒体可出现功能障碍,具体表现为线粒体膜电位降低、ATP合成减少、胞内Ca2+超载、线粒体通透性转变孔道(mitochondrial permeability transition pore,mPTP)开放、ROS剧增、细胞色素C及凋亡诱导因子等释放,caspase激活,引起相关信号通路介导细胞损伤,最后导致细胞凋亡或坏死甚至可能发生焦亡[10]。
2 糖尿病血管并发症
2.1 主要特点 高血糖被认为是糖尿病性血管疾病及其他并发症的初始病因,长期高血糖可导致糖尿病及其并发症,其中又以血管并发症为主。糖尿病血管并发症分为大血管并发症及微血管并发症,大血管并发症包括糖尿病足、外周神经病变等,微血管并发症包括糖尿病性眼病、视网膜病变、白内障、勃起功能障碍等[11-12]。内皮功能障碍是糖尿病血管并发症早期的关键病理生理步骤(图1)[13]。内皮细胞排列在血管腔内,是血管内稳态的关键调节器,调节各种分子向动脉内膜的运输,同时也产生不同的分子来调节血管功能。糖尿病患者的血糖异常可损害血管内皮功能[14],高血糖的糖毒性作用可导致血管内皮细胞功能发生障碍,如一氧化氮(nitric oxide,NO)活性下降,黏附因子上调,活性氧(reactive oxygen species,ROS)增多等。尽管糖尿病血管内皮功能的调节机制仍未完全阐明,但研究证实,线粒体功能紊乱在糖尿病血管疾病的早期发病机制及最终临床表现中具有独特的作用,内皮线粒体在局部及全身水平上对调节血管内皮功能起关键作用[15-17]。有文献报道,在离体血管中线粒体复合物1抑制剂鱼藤酮可明显损害乙酰胆碱对血管张力的内皮依赖性调节作用[18]。另有研究证实,高血糖能够激活线粒体介导的凋亡,导致内皮细胞死亡[19],人微血管内皮细胞暴露在高糖状态24 h,线粒体的呼吸功能可受到抑制,从而证实高糖能够造成线粒体功能障碍[18]。Paneni等[20]发现,高糖可促进Pin1(参与

图1 糖尿病血管并发症的发病机制Fig.1 Pathogenesis of diabetic vascular complications
p66ShC及P53氧化蛋白的线粒体定位)表达,导致线粒功能紊乱与细胞色素C释放,而敲除Pin1基因可使得糖尿病小鼠血管的内皮依赖性舒张功能得到恢复,核因子κB(nuclear factor kappa-B,NF-κB)、血管细胞黏附分子1(vascular cell adhesion molecule-1,VCAM-1)、细胞间黏附分子-1(intercellular cell adhesion molecule-1,ICAM-1)及单核细胞趋化蛋白-1(monocyte chemoattractant protein-1,MCP-1)的表达受到抑制,证实了线粒体与血管功能障碍存在一定关系。
2.2 线粒体功能障碍的影响 大量关于糖尿病发病机制的研究开始关注亚细胞器,尤其是近几年线粒体及其功能的改变在代谢性疾病、癌症中的研究已成为热点。线粒体在糖尿病及其各类血管并发症中发挥重要作用,在错综复杂的致病机制中均伴随线粒体功能障碍。在糖尿病病理状况下,ROS大量生成、NADPH导致的氧化应激、促炎因子大量释放导致炎症反应[21]。以上机制单一或相互作用致线粒体明显肿胀、外膜破裂、线粒体DNA损伤、呼吸链抑制等造成能量代谢障碍,从而导致一系列相互作用的损伤过程,甚至是级联放大损伤[22]。
2.2.1 线粒体氧化应激 尽管ROS可以在多种信号级联的多个细胞室中产生,但大多数的ROS是由线粒体能量代谢产生的。糖尿病的持续高血糖状态可使ROS增加,而ROS大量生成可导致糖尿病的微血管及大血管并发症[23-24]。高血糖可通过3种方式(引起呼吸链电子传递受阻、ROS暴发式产生、损伤抗氧化系统)导致ROS相对增多,介导线粒体氧化应激,损伤线粒体,引起线粒体功能障碍[25]。线粒体ROS的产生不仅减少了NO的生物利用度,还驱动了线粒体诱导的凋亡[26]。但Merdzo等[27]发现,糖尿病大鼠脑动脉存在ROS生成增加的现象,但线粒体呼吸作用及蛋白水平维持正常,意味着可能存在相关的补偿机制。另外,ROS积累及其介导的炎性反应为糖尿病血管表型的主要特征[2,28]。Brownlee等[29]的“糖尿病并发症的共同机制”学说认为不管是糖尿病大血管还是微血管并发症,氧化应激为其共同的发病机制。高血糖引起线粒体内超氧阴离子()生成过多,导致多元醇通路激活、氨基己糖途径激活、蛋白激酶C(protein kinase C,PKC)途径活化、晚期糖基化终末产物(advanced glycation end products,AGEs)形成,引起细胞功能紊乱,最终引起糖尿病血管病变,如糖尿病的大血管并发症[21]。Ceriello等[30]提出“共同土壤学说”:糖尿病、胰岛素抵抗及心血管疾病的共同发病基础是氧化应激。来源于线粒体的ROS激活诱导糖尿病并发症,并与糖尿病血管病变的相关机制相互作用,加剧或放大ROS介导的氧化应激和(或)炎症反应。体外实验发现,高血糖可以刺激内皮细胞产生ROS并增加NF-κB,激活PKC及AGEs[31-32]。这也佐证了高血糖导致细胞内ROS生成过量或清除功能受损从而引起了氧化应激[25,33](图2)。还有研究表明,高血糖通过活化还原型辅酶Ⅱ(nicotinamide adenine dinucleotide phosphate,NADPH)刺激下游的ROS大量生成,引起氧化应激,最终增加罹患糖尿病血管疾病的风险[34]。有研究采用链脲佐菌素(STZ)建立糖尿病小鼠模型,引起ROS增多诱导氧化应激,使得M2型瞬时受体电位(transient receptor potential melastatin,TRPM2)及瞬时电位香草酸受体1(transient receptor potential vanilloid 1,TRPV1)过表达,导致Ca2+超载,出现神经疼痛及海马损伤,但给予褪黑素、硒后可有不同程度改善[35]。
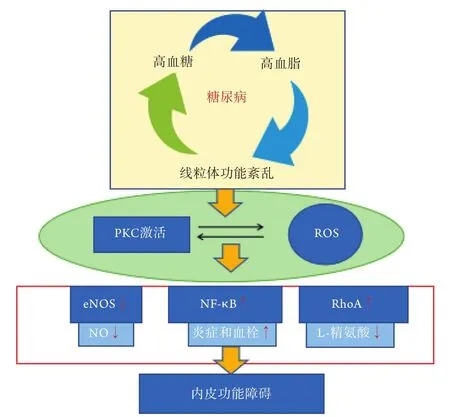
图2 氧化应激介导的糖尿病血管内皮功能障碍Fig.2 Oxidative stress mediated vascular endothelial dysfunction in diabetes
2.2.2 线粒体钙紊乱 线粒体Ca2+超载是线粒体功能障碍的主要原因。内质网储存Ca2+、线粒体摄取Ca2+,二者通过线粒体内质网相关膜及基质共同维持细胞内钙稳态。线粒体膜电位增加Ca2+摄取,膜电位降低,线粒体Ca2+摄取减少、外流增加[36-37],ROS、mPTP开放[38],线粒体DNA损伤等造成线粒体Ca2+紊乱[39]。
线粒体Ca2+超载是糖尿病小鼠内皮细胞凋亡的原因之一。有研究发现,糖尿病小鼠的冠状动脉内皮细胞中Ca2+浓度明显升高,导致糖尿病内皮细胞ROS明显升高[40]。线粒体Ca2+超载是高血糖期间线粒体诱导内皮细胞凋亡的关键因素,逆转糖尿病小鼠冠状动脉内皮细胞线粒体Ca2+水平可改善糖尿病冠状动脉微血管病变,表明预防线粒体Ca2+超载是减少糖尿病血管内皮细胞凋亡的潜在治疗靶点[40-41]。线粒体Ca2+外通道蛋白及转运蛋白改变也是引起线粒体Ca2+超载的原因,如TRPV蛋白通道、Na+/Ca2+转运蛋白损伤引起线粒体Ca2+超载[42]。Ca2+紊乱导致糖尿病鼠的心功能发生障碍,引起钙通道蛋白兰尼碱受体(ryanodine receptors,RyR2)、心肌肌质网Ca2+-ATP酶(sarocplasmic reticulum Ca2+-ATPase,SERCA)、钠钙离子交换蛋白1(Na+/Ca2+exchanger,NCX1)随糖尿病心肌病时间的延长而增加[43]。氧化应激也可以通过增加钙通道蛋白RyR2的氧化引起线粒体Ca2+紊乱[44]。再如G-蛋白偶联受体或者受体络氨酸激酶可引起Ca2+通道激活,Ca2+大量内流,诱导糖尿病血管并发症[45]。Gan等[46]研究发现,20(S)-原人参二醇可抑制细胞外Ca2+通过受体操纵钙通道(receptor-operated calcium channels,ROCC)和电压依赖性钙通道(voltage-dependent calcium channels,VDCC)流入引起的血管收缩,同时使钙激活性钾通道开放,从而导致血管舒张。通过增加电压依赖性阴离子通道(voltage-dependent anion channel,VDAC)的内源性抑制剂己糖激酶2的表达,影响Ca2+/ROS水平,从而逆转糖尿病大鼠的冠状动脉微血管病变[42]。
基于上述病理改变,一系列药物的开发取得了较好进展。如苦参碱可通过抑制RyR2减少Ca2+内流,改善AGEs引起的糖尿病心肌病对心肌功能的影响[32]。糖尿病合并心肌缺血的小鼠通过活化Na+/Ca2+等交换器引起Ca2+超载,加剧糖尿病介导的心肌缺血[47]。
2.2.3 线粒体通透性转变孔道开放 mPTP开放可导致线粒体功能发生障碍,甚至细胞死亡。诱发线粒体mPTP开放的因素很多,如线粒体内ATP减少、Ca2+超载、ROS产生、游离脂肪酸增多等,最重要的是氧化应激及Ca2+超载[48-49]。糖尿病合并缺血时,VDAC等通道蛋白通透性增加,mPTP开放导致呼吸链复合物1-5释放,大量H+回流;内膜去极化导致电位剧增至140 mV,引起氧化磷酸化完全解偶联,ATP合成受阻;基质外流,谷胱甘肽耗竭,大量生成;线粒体肿胀,外膜破裂,释放大量细胞色素C、ROS等,caspase-3成熟释放引起线粒体途径细胞凋亡[50-52]。
2.2.4 线粒体动力学变化 在生理情况下,线粒体分裂及融合的平衡维持着线粒体的正常功能,修复受损的线粒体及阻止受损线粒体的过度自噬。但在病理条件下,线粒体分裂融合的平衡失调导致线粒体功能障碍。高糖通过上调分裂相关蛋白如线粒体动力相关蛋白(dynamin-related protein 1,Drp1)及线粒体分裂蛋白(fission protein 1,Fis1),诱导心肌细胞线粒体过度分裂,出现细胞凋亡及细胞周期停滞[53]。干预Fis1、Drp1在糖尿病血管并发症引起的线粒体动力学失衡中至关重要,可作为糖尿病血管并发症线粒体功能障碍的干预靶点[54-55]。
细胞可根据细胞内信号及细胞外微环境刺激改变线粒体的分裂融合平衡。在细胞周期的G2/M期,线粒体分裂增加,以保证细胞分裂时子细胞之间的线粒体分离;分裂又能促进细胞色素C释放到胞质内,催化细胞内源性凋亡(图3)[56]。沉默Mfn1基因可下调线粒体对血管内皮生长因子及蛋白激酶B依赖的内皮NO合酶效应,抑制线粒体融合后的血管生成[57]。线粒体的分裂融合还与线粒体膜电位相关,糖尿病应激及高糖可通过降低线粒体膜电位导致的内皮祖细胞凋亡[58],而膜电位的丢失又会进一步增加ROS的生成,加剧线粒体的功能障碍。

图3 线粒体的分裂-融合Fig.3 Mitochondrial division-fusion
2.3 改善线粒体功能的治疗作用 由于线粒体参与了不同疾病的发生,人们据此开发了潜在的治疗药物,其主要作用机制是防止氧化应激、改善代谢功能、调节钙稳态、改善线粒体的生理状态。近年来的研究多将胰岛素生长因子-1(insulin-like growth factor-1,IGF-1)与线粒体的功能及氧化应激联系起来,用于改善线粒体的功能障碍。Puche等[59]发现,IGF-1的作用与线粒体保护密切相关,可使自由基产生、氧化损伤及细胞凋亡减少,同时增加ATP生成。经IGF-1处理的人脐静脉内皮细胞表现为线粒体膜电位保守、线粒体细胞色素C释放减少及caspase-3活性降低,而这些恰恰与糖尿病血管并发症的生理病理机制相似。Sádaba等[60]提出,用小剂量IGF-1治疗线粒体功能紊乱可能是恢复正常线粒体功能的一种有效方法。针对线粒体的分裂融合,可使用线粒体分裂抑制剂(Mdivi-1)改善2型糖尿病患者的动脉内皮舒张功能[61]。此外,在血管紧张素Ⅱ(AngⅡ)导致的内皮功能障碍中,Mdivi-1逆转了AngⅡ的作用,保护了内皮细胞,同时减少了ROS的产生[62]。在中药开发方面,有研究报道,白藜芦醇可改善血管功能,其机制是介导沉默信息调节因子1(silent information regulator 1,SITR1)激活调节过氧化物酶体增殖物激活受体γ共激活因子1α(peroxisome proliferator-activated receptor γ coactivator-1,PGC-1α)脱乙酰,从而改善线粒体的蛋白合成及生理功能[63]。另有研究发现,白藜芦醇在抗糖尿病的多重作用中也可抑制Drp1介导的线粒体分裂[64]。
3 总 结
糖尿病及胰岛素抵抗是最常见的代谢紊乱[65]。冠心病、卒中等心脑血管并发症是糖尿病患者死亡的主要原因[22]。糖尿病合并心血管疾病患者的病死率约70%[66]。糖尿病发病机制复杂且不明确,线粒体功能障碍与糖尿病及其血管并发症密切相关,线粒体导致的氧化应激、Ca2+紊乱、mPTP开放等相互作用于不同的糖尿病血管并发症。因此,积极寻找相关靶点的抑制剂或分子标志物可以为临床治疗提供实验依据,也是糖尿病及其血管并发症新的研究方向。
