并发杜氏肌营养不良症的复合型甘油激酶缺乏症一例并文献复习
2020-04-27刘靖薇江文文杨京华林晓红
刘靖薇 江文文 杨京华 林晓红

【摘要】目的 分析并发杜氏肌营养不良症(DMD)的复合型甘油激酶缺乏症(CGKD)的诊治要点。方法 报道1例并发DMD的CGKD病例,并以“复合型甘油激酶缺乏症”和“杜氏肌营养不良”为关键词(包括中英文)在PubMed和中国期刊全文数据库(CNKI)进行检索,收集并分析相关患者的资料。结果 该例患儿以双下肢乏力为主要表现,伴皮肤色素沉着,尿液有机酸综合分析示甘油显著升高,促肾上腺皮质激素升高而血皮质醇降低,经基因检测明确DMD及CGKD的诊断,予氢化可的松口服激素替代治疗与其他对症治疗,患儿病情好转且稳定。检索文献共收集到20篇相关的病例报道,小年龄CGKD伴DMD患儿的肌营养不良相关症状不明显,多表现为无原因肌酶升高,随着年龄增长患儿逐渐出现肌肉乏力症状,预后差。CGKD 目前尚无有效根治方法,以氢化可的松替代治疗、低脂饮食以及康复治疗等对症治疗为主。结论 伴发DMD的CGKD患儿预后差,因此产前诊断更为重要,临床医师需提高认识,及时行基因检测以提高临床确诊率。
【关键词】甘油激酶缺乏症;先天性肾上腺发育不良;杜氏肌营养不良症;儿童
【Abstract】Objective To analyze the diagnosis and treatment of complex glycerol kinase deficiency (CGKD) complicated with Duchenne muscular dystrophy (DMD). Methods One case of CGKD complicated with DMD was reported. Literature review was conducted from PubMed and CNKI by using the keywords of ‘complex glycerol kinase deficiency and ‘Duchenne muscular dystrophy both in Chinese and English. The data of the searched patients were collected and analyzed. Results The main manifestation of the patient was hypodynamia of bilateral lower limbs, accompanied by skin pigmentation. Comprehensive analysis of urine organic acid showed that the glycerol level was significantly increased, adrenocorticotropic hormone level was increased whereas blood cortisol level was decreased. The diagnosis of CGKD complicated with DMD was confirmed by gene assay. The patients condition was improved and stable after oral hydrocortisone hormone replacement therapy and other symptomatic treatments. A total of 20 case reports were retrieved. The muscular dystrophy-related symptoms were not obvious in CGKD complicated with DMD children of young age. A majority of them presented with increased kinase level with unknown causes, and gradually presented with muscular weakness symptoms over age. The clinical efficacy in the treatment of CGKD complicated with DMD is worse. At present, there is no effective radical treatment for CGKD. Hydrocortisone replacement therapy, low-fat diet and rehabilitation therapy are adopted as the main symptomatic treatments. Conclusions The clinical prognosis of CGKD children complicated with DMD is poor. Therefore, prenatal diagnosis is of more significance. Clinicians should improve their understanding of CGKD complicated with DMD and deliver gene detection timely to enhance the clinical diagnosis rate.
【Key words】Glycerol kinase deficiency;Congenital adrenal dysplasia;
Duchenne muscular dystrophy;Children
甘油激酶缺乏癥(GKD)具体类型包括单纯型和复合型,其中复合型GKD(CGKD)因其同时出现数个单基因缺陷病的症候群,故极易被误诊为其中一种单基因缺陷病[1]。为提高临床的确诊率,现报道我院收治的并发杜氏肌营养不良症(DMD)的CGKD患儿1例,并检索相关文献进行分析。
对象与方法
一、1例并发DMD的CGKD患儿临床资料的收集
我科于2019年6月20日收治1例以肢体乏力为首要症状的并发DMD的CGKD患儿,收集并分析其病史、各项检查、治疗及转归等资料。
二、文献检索
以“复合型甘油激酶缺乏症”和“杜氏肌营养不良”为关键词(包括中英文)对PubMed和中国期刊全文数据库(CNKI)中截至2019年12月收录的论文进行检索,收集并分析有DMD表现的CGKD患者的资料。
结果
一、1例并发DMD的CGKD患儿的病历资料
1.主诉及病史
患儿男,3岁,因双下肢乏力1年余于2019年6月20日入广东省中医院。患儿入院时以双下肢乏力、咳嗽为主要症状。患儿为第2胎,足月剖腹产出,出生时无产伤及窒息史,出生后母乳喂养,按时添加辅食,出生时肤色正常,约6月龄后皮肤颜色逐渐加深,生长发育缓慢,10月龄抬头,1岁坐起,1岁余可在扶持下走路,2岁开始独立走路后有双下肢乏力症状,家长未予重视。患儿哥哥(同母异父)会行走后亦被发现存在双下肢乏力症状,但未接受系统诊治,4岁余时因被狗咬而感染离世。患儿父系及母系中其他成员均无相关症状。2016年初,患儿因发热至当地医院就诊,查心肌酶升高,曾有抽搐伴高钾、低钠血症(具体不详),未接受系统诊治;患儿于2018年5月与6月及2019年1月与5月因反复肺部感染在当地医院住院,伴心肌酶升高及肝功能异常,尿液有机酸综合分析示甘油显著升高,促肾上腺皮质激素392.4 pmol/L,游离睾酮0.51 pg/ml,雄烯二酮<0.3 ng/ml,遗传代谢病氨基酸和酰基肉碱无明显异常,诊断为∶①重症肺炎;②可疑进行性肌营养不良;③心肌损害;④肝功能异常;⑤可疑甘油酸代谢紊乱,予抗感染等对症治疗后,患儿病情好转并出院,但双下肢仍乏力。3 d前患儿无明显诱因出现咳嗽,遂到我科住院治疗。
2.体格检查、实验室及辅助检查
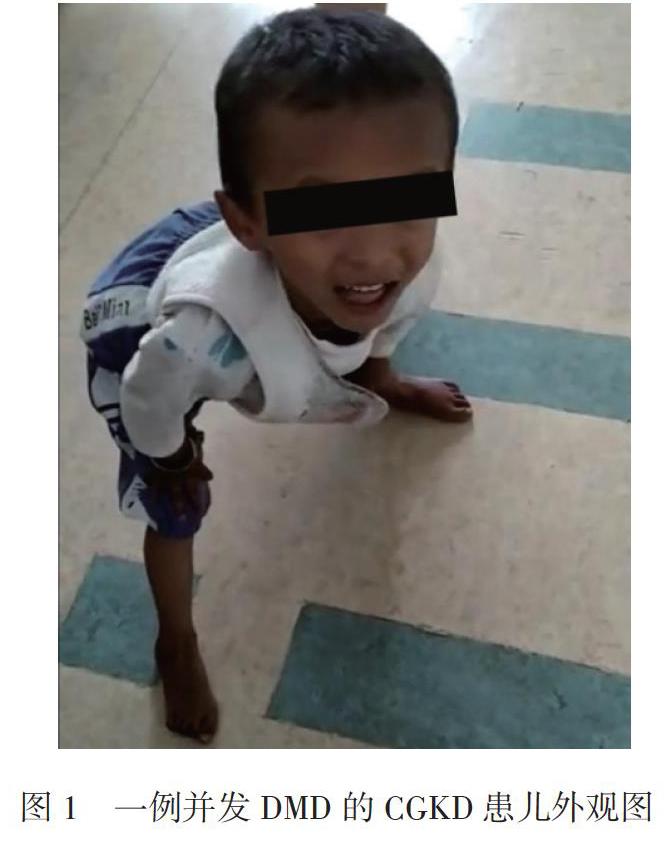
体格检查:神志清晰,精神尚可,营养欠佳,形体偏瘦,构音欠清,行走欠稳似鸭步,查体欠合作。全身皮肤黝黑,口唇、牙龈色暗,乳晕、阴囊色素沉着,见图1。双腓肠肌无假性肥大,触之僵硬;四肢肌张力正常,四肢肌力正常;Gower征(+),四肢腱反射稍减弱,四肢深、浅感觉正常。
2019年6月20日肝功能:ALT 145 U/L,AST 138 U/L;心肌酶:肌酸激酶7856 U/L,肌酸激酶同工酶382.3 U/L,乳酸脱氢酶1099 U/L;促肾上腺皮质激素> 440.4 pmol/L。考虑不排除患儿有DMD、肾上腺皮质功能减退、先天性代谢性疾病等,遂继续完善相关检查。2019年6月24日血皮质醇(上午7 ~ 9时) < 13.8 nmol/L;甲状腺功能:T3 2.83 nmo1/L,FT3 7.44 pmo1/L,TSH 10.14 IU/L;性激素:卵泡刺激素0.14 U/L,黄体生成素0.20 U/L,
催乳素618.00 mIU/L,雄二醇 < 18.35 pmol/L,睾酮 < 0.09 nmol/L;血脂:甘油三酯4.58 mmo1/L,
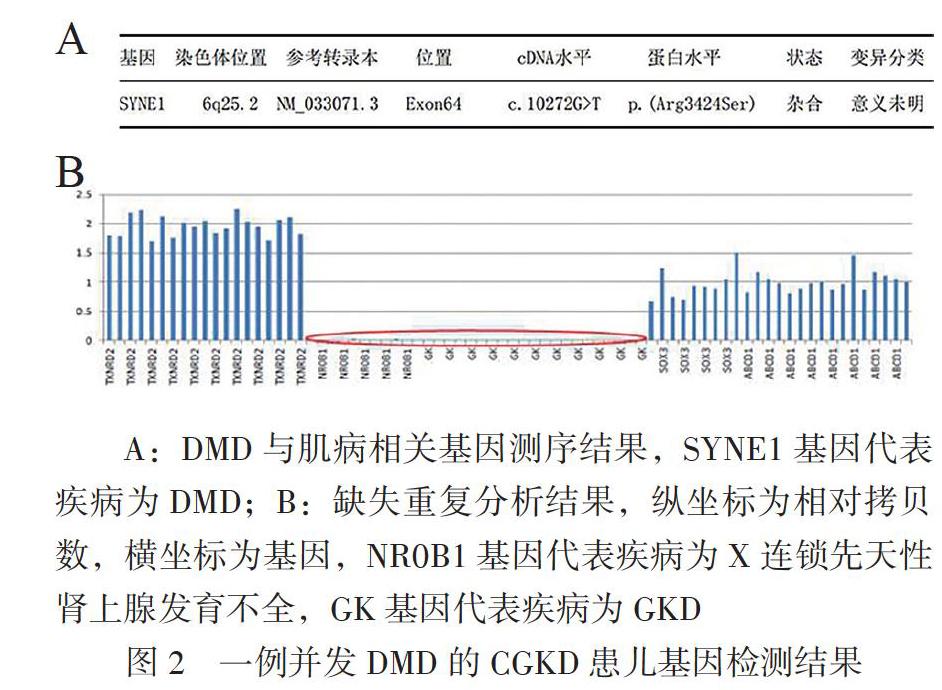
总胆固醇2.30 mmo1/L,HDL-C 0.5mmol/L,载脂蛋白A1 0.77 g/L,载脂蛋白B 0.56 g/L。基因检测:①高通量二代测序方法[多重连接探针扩增技术(MLPA)]检测到受检者SYNE1基因(NM_00 4006.2)56-79号外显子半合子缺失,其余外显子未见异常,见图2A,该变异为缺失突变,预计会使所编码蛋白质发生紊乱,进而丧失其正常功能而致病;②高通量测序技术,又称“下一代”测序技术(NGS),结果显示该样本Xp21.2(chrX:30322676-30746913)区域发生424 Kb缺失,为致病性变异,见图2B。
二、治疗与转归
患儿确诊为CGKD依据如下:①患儿出现双下肢乏力、色素沉着、生长发育迟缓等症状;②肝功能异常,心肌酶升高,促肾上腺皮质激素增高伴皮质醇降低,甘油三酯增高,既往曾有抽搐伴高钾、低钠血症;③患儿X 染色体Xp21.2(chrX:30322676-30746913)区域发生424 Kb缺失,DMD基因(NM_004006.2)56-79号外显子半合子缺失。根据患儿病史、症状及辅助检查结果,确诊为CGKD。治疗上,予口服氢化可的松(晨起5 mg,下午2.5 mg)替代治疗、复方甘草酸苷片护肝,限制动物油等饱和脂肪酸摄入,并告知家长患儿肾上腺皮质功能低下,易在感染、发热、外伤等应激状态下出现肾上腺皮质危象,预后差,家长表示知情理解。随访情况如下:患儿于2019年6月27日出院,后因肺炎入住当地医院ICU,经系统治疗后1周出院,随访至2020年1月,患儿一般情况稳定,仍有双下肢乏力,皮肤黝黑症状改善。
三、文献检索结果
检索数据库后共收集到20篇相关病例报道[2-21]。最早被报道的是1983年的1例荷兰患儿[22]。发病年龄为新生儿期(具体不详),表现为DMD、生长障碍、智力迟鈍、肾上腺皮质功能不全,予长达半年的激素治疗,后未续予激素替代治疗,幼年时期出现过1次癫痫。综合相关文献报道,DMD症状表现方面,小年龄者DMD相关症状不明显,多发现无原因肌酶明显升高,随着年龄增长运动发育迟缓,年龄大者多出现肌肉乏力症状。现总结2015 ~ 2019年9例并发DMD的CGKD的典型病例资料,见表1。
讨论
DMD发病者多数为男性患儿,可有双下肢乏力,Gower征(+)等临床表现,实验室检查提示心肌酶、转氨酶升高,部分患儿可见生长发育迟缓,本例患儿即有上述表现,且其家族中同母异父的哥哥亦有运动发育迟缓的情况,但其父亲兄弟的孩子、母亲家系其他成员无相关症状,考虑患病与伴X染色体基因隐性遗传有关,符合DMD表现。然而DMD不能解释患儿出现的皮肤色素沉着、促肾上腺皮质激素明显升高、既往反复多次感染、高甘油尿症等临床情况。追溯患儿病史:患儿反复多次因高热于当地医院住院,并出现低血钠、高血钾等急性失盐表现,住院期间曾出现2次抽搐,符合肾上腺皮质功能减退的临床表现,应当完善皮质醇水平、性激素、甲状腺激素等检查以鉴别原发性还是继发性。然而该病不能解释高甘油尿症,双下肢乏力等症状。因此上述单一疾病无法解释患儿的所有临床特点,需要注意合并罕见疾病的情况。
GKD是一種罕见的遗传代谢性疾病,与X染色体隐性遗传相关,具体类型包括单纯型和复合型。CGKD是Xp21.3-21.2区域基因片段缺失突变导致的数个单基因缺陷病,其临床表现随缺失片段多少而变化,常见受累的是甘油激酶基因位点相邻的基因位点,如DMD、先天性肾上腺发育不良(AHC)、鸟氨酸氨甲酰基转移酶缺乏、视网膜色素变性、慢性肉芽肿等 ,其中由于GKD基因位点距离最近的是AHC和DMD基因位点, 因而复合型以AHC-GKD-DMD最为常见,故常被误诊为其中一种疾病[16]。以下以AHC-GKD-DMD为例概述其主要临床特征。
CGKD的临床表现取决于GK基因邻近缺失片段多少,如SYNE1基因缺失患儿会出现血清各种酶谱升高,如心肌酶、转氨酶等,伴或不伴肌无力等[23-25]。NR0B1基因缺失患儿常出现原发性肾上腺功能不足(PAI)和严重失盐的临床表现,包括反复感染、进行性皮肤色素沉着等,部分患儿可在青春期出现促性腺激素减少,且需要与其他先天性肾上腺功能低下疾病相鉴别[25-28]。维持机体正常代谢所必需的特殊酶相关基因缺失会导致遗传代谢病[29]。GK基因缺失患儿会出现甘油激酶缺乏所致的高甘油血症及高甘油尿症、低血糖甚至抽搐等临床表现的遗传代谢病。此外,相关文献报道若同时发生基因大片段缺失,患儿会出现明显的精神运动发育迟缓[30]。精神运动发育迟缓为多因素的结果,包括PAI未及时诊疗导致电解质紊乱,或GKD婴幼儿期发作性低血糖,引起神经系统损伤,DMD基因或IL1RAPL基因突变或缺失与之相关[16]。国外病例报道示,CGKD还可表现为身材矮小、严重发育迟缓和畸形的特征[31]。
体格检查方面,CGKD比较典型的是AHC基因缺失所致的进行性皮肤色素沉着,全身皮肤(皮肤皱褶尤甚)及黏膜色素沉着;DMD基因缺失所致的Gower征(+),如伴有肝功能异常严重者,可伴黄疸。若临床表现及体格检查有比较典型的表现,可以行尿液有机酸综合分析以了解有无高甘油尿症,检测促肾上腺皮质激素及皮质醇以了解肾上腺皮质减退的情况,而最重要的是分子遗传学分析,有助于确定基因缺失的范围。
目前尚无有效根治CGKD的方法,主要以氢化可的松替代治疗、低脂饮食以及康复治疗等对症治疗为主。此类患儿预后差,因此产前诊断更为重要[12]。结合本例患儿,其以双下肢乏力为主要症状,伴皮肤进行性色素沉着,反复感染,既往当地医院诊断为可疑进行性肌营养不良,仔细分析其病史及其临床表现,可以发现其不是单纯的肌病,进一步完善相关辅助检查得以明确诊断。目前已有大约100例CGKD患者被报道,其中有17例因肾上腺功能障碍未被发现或处理不当在新生儿期或儿童期死亡[13]。因CGKD常以AI为首发症状,故临床上遇到肾上腺皮质功能低下的患儿,尤其是伴肌酶升高、血脂异常者,应进一步完善尿有机酸分析以及遗传学检查,以降低误诊率。
参 考 文 献
[1] Montoya-Williams D,Mowitz M. Cholestasis and hepatic iron deposition in an infant with complex glycerol kinase deficiency. Pediatrics, 2017, 140(1):e20161479.
[2] Bartley JA, Patil S, Davenport S, Goldstein D, Pickens J. Duchenne muscular dystrophy, glycerol kinase deficiency, and adrenal insufficiency associated with Xp21 interstitial deletion. J Pediatr, 1986,108(2):189-192.
[3] Sanz-Ruiz I, Bretón-Martínez JR, Del Castillo-Villaescusa C, Cásanovas-Martínez A, Martínez-Castellano F, Millán-Salvador JM, Hernández-Marco R, Codo?er-Franch P. Contiguous gene deletion syndrome in Xp21: an unusual form of presentation. Rev Neurol, 2009,49(9):472-474.
[4] Pantoja-Martínez J, Martínez-Castellano F, Tarazona-Casany I, Buesa-Ibá?ez E, Ardid-Encinar M, Esparza-Sánchez MA, Bonet-Arzo J. Contiguous gene deletion syndrome in Xp21: The association between glycerol kinase deficiency, congenital suprarenal hypoplasia and Duchennes muscular dystrophy. Rev Neurol, 2007, 44(10):606-609.
[5] Amato AA . Duchenne muscular dystrophy and glycerol kinase deficiency. J Clin Neuromuscul Dis, 2000, 1(4):191.
[6] Guggenheim MA, McCabe ER, Roig M, Goodman SI, Lum GM, Bullen WW, Ringel SP. Glycerol kinase deficiency with neuromuscular, skeletal, and adrenal abnormalities. Ann Neurol, 1980, 7(5):441-449.
[7] Heide S, Afenjar A, Edery P, Sanlaville D, Keren B, Rouen A, Lavillaureix A, Hyon C, Doummar D, Siffroi JP, Chantot-Bastaraud S.Xp21 deletion in female patients with intellectual disability: two new cases and a review of the literature. Eur J Med Genet, 2015, 58(6-7):341-345.
[8] Wikiera B, Jakubiak A, Zimowski J, Noczyńska A, Smigiel R. Complex glycerol kinase deficiency-X-linked contiguous gene syndrome involving congenital adrenal hypoplasia, glycerol kinase deficiency, muscular Duchenne dystrophy and intellectual disability (IL1RAPL gene deletion). Pediatr Endocrinol Diabetes Metab,2012,18(4):153-157.
[7] Stuhrmann M, Heilbronner H, Reis A, Wegner RD, Fischer P, Schmidtke J. Characterisation of a Xp21 microdeletion syndrome in a 2-year-old boy with muscular dystrophy, glycerol kinase deficiency and adrenal hypoplasia congenita. Hum Genet, 1991, 86(4):414-415.
[10] 李秀珍,劉丽,梅慧芬.儿童复合型甘油激酶缺乏症.中国当代儿科杂志,2007,9(5):441-444.
[11] 麻宏伟,王志超,王华,张海娟. Xp21邻近基因缺失综合征1例.中国实用儿科杂志,2002,17(3):159-160.
[12] 田飞,张颖,王彩霞,张淑萍,陈志红. Xp21邻近基因缺失综合征1例.中国实用儿科杂志,2002,17(3):159-160.
[13] 王旭,吴迪,方方,姜敏. Xp21临近基因缺失综合征6例临床和遗传学研究.中国实用儿科杂志,2015,30(7):61-65.
[14] 郑章乾,吴冰冰,章淼滢,陆炜,罗飞宏. 反复纳差伴皮肤色素沉着2月余. 中国当代儿科杂志,2017,19(8):926-929.
[15] 刘晖,王子敬,郑启安,陈燕玲,陈悠涛,廖醒.复合型甘油激酶缺乏症1例报告.临床儿科杂志,2012,30(3):286-290 .
[16] 范瑞,张一宁,李小平,陆飞宇,杜红伟.复合型甘油激酶缺乏症2例报告并文献复习.临床儿科杂志,2018,36(3):197-201.
[17] 杨楠,韩连书,顾学范,鲍一笑. 腹泻 皮肤色素沉着 阴囊发育不良.中国实用儿科杂志,2011,26(12):946-947.
[18] 刘玉凤,贾雁平. 甘油酸激酶缺乏症一例.中华新生儿科杂志, 2015,30(2):143.
[19] Wikiera B, Jakubiak A, Zimowski J, Noczyńska A, Smigiel R. Complex glycerol kinase deficiency - X-linked contiguous gene syndrome involving congenital adrenal hypoplasia, glycerol kinase deficiency, muscular Duchenne dystrophy and intellectual disability (IL1RAPL gene deletion). Pediatr Endocrinol Diabetes Metab, 2012, 18(4):153-157.
[20] Chelly J, Marlhens F, Dutrillaux B, Van Ommen GJ, Lambert M, Haioun B, Boissinot G, Fardeau M, Kaplan JC. Deletion proximal to DXS68 locus (L1 probe site) in a boy with Duchenne muscular dystrophy, glycerol kinase deficiency, and adrenal hypoplasia. Hum Genet, 1988 ,78(3):222-227.
[21] Korkut S, Osman Ba?tu?, Raygada M, Hatipo?lu N, Kurto?lu S, Kendirci M, Lyssikatos C, Stratakis CA. Complex glycerol kinase deficiency and adrenocortical insufficiency in two neon-ates. J Clin Res Pediatr Endocrinol, 2016, 8(4):468-471.
[22] Renier WO, Nabben FA, Hustinx TW, Veerkamp JH, Otten BJ, Ter Laak HJ, Ter Haar BG, Gabre?ls FJ. Congenital adrenal hypoplasia, progressive muscular dystrophy, and severe mental retardation, in association with glycerol kinase deficiency, in male sibs. Clin Genet,1983,24(4):243-251.
[23] 杨茹莱.杜氏肌营养不良临床特点及诊治进展. 中国儿童保健杂志,2018,26(3):233-235.
[24] 王红,闫丹丹,李丽,赵学良,王叶. 以心肌酶异常起病的杜氏肌营养不良1例.中国实验诊断学,2019,23(8):1353-1354.
[25] 宋宁忆.因肝酶升高就诊的杜氏肌营养不良2例报告并文献复习.中国社区医师,2016,32(35):173-175.
[26] 楊露露,董治亚. 原发性肾上腺皮质功能减退分子发病机制研究进展.中国实用儿科杂志,2016,31(6):477-480.
[27] 李嫔.原发性肾上腺皮质功能减退临床诊断.中国实用儿科杂志,2016,31(6):414-418.
[28] Serbis A, Tsinopoulou VR, Mouzaki K, Kotanidou EP, Giza S, Galli-Tsinopoulou A. Testicular microlithiasis in a boy with X-linked adrenal hypoplasia congenita. Ann Pediatr Endocrinol Metab,2018,23(3):162-165.
[29] 董红,欧榕琼,欧阳颖. 遗传代谢病类婴儿肝炎综合征的临床分析.新医学,2015,46(10):60-63.
[30] Heide S, Afenjar A, Edery P, Sanlaville D, Keren B, Rouen A, Lavillaureix A, Hyon C, Doummar D, Siffroi JP, Chantot-Bastaraud S. Xp21 deletion in female patients with intellectual disability: two new cases and a review of the literature. Eur J Med Genet, 2015, 58(6-7):341-345.
[31] Almontashiri NAM, Berry GT, Majzoub J, Peake RWA.Abnormal glycerol metabolism in a child with global develop-mental delay, adrenal insufficiency, and intellectual disability. Clin Chem,2018,64(12):1785-1787.
(收稿日期:2020-01-01)
(本文编辑:洪悦民)
