NLRP3基因变异致听力损失的临床特征及研究进展
2020-04-27吴侃王浩然吴玉森兰兰熊芬谢林怡王洪阳关静王大勇王秋菊
吴侃 王浩然 吴玉森 兰兰 熊芬 谢林怡 王洪阳 关静 王大勇 王秋菊
中国人民解放军总医院耳鼻咽喉头颈外科医学部,国家耳鼻咽喉疾病临床医学研究中心,解放军耳鼻咽喉研究所,聋病教育部重点实验室;聋病防治北京市重点实验室(北京100853)
双侧迟发型听力损失是一种特殊的听力损失类型,其预后较差,临床表现多样,病因复杂,血管、肿瘤、中毒、传染病、全身伴随疾病等因素可能与之相关[1]。此外,免疫因素和遗传因素也不容忽视。近年来,NLRP3基因变异导致的双侧迟发型听力损失以其特殊的炎性反应临床表现和显著的遗传学倾向受到越来越多的关注。
NLRP3基因(OMIM:*606416)全称NLR family pyrin domain containing 3,编码cryopyrin样蛋白参与NLRP3炎性小体复合物的构成,后者在炎症、免疫反应和凋亡的调控中发挥作用。该基因变异引起IL-1β的生成过多,从而启动免疫级联反应、一系列炎性因子的释放和细胞凋亡[2],导致冷冻蛋白相关周期综合征(cryopyrin-associated periodic syndrome,CAPS)。CAPS根据症状轻重分为3种亚型,他们之间互相重叠,构成连续的疾病谱,大约42%~89%的患者可以出现进展性感音神经性听力损失(sensorineural hearing loss,SNHL)[3-5]。NLRP3 也被认为是DFNA34的致病基因[6]。本研究发现3例特殊临床表现的双侧迟发型听力损失病例,通过临床资料的分析和新一代测序技术的检测,明确为NLRP3基因变异相关性耳聋,并且回顾复习了52篇相关文献,以寻求有效的药物治疗方案。
1 材料与方法
1.1 研究对象
病例1为2016年3月首诊于我院耳内科门诊的双侧混合性听力损失患者,出现反复发热、皮疹、炎性标志物增高等特殊临床表现,通过新一代测序技术明确为NLRP3基因变异相关性耳聋,其后回顾双侧迟发型听力下降患者基因检测结果,又发现2例携带该基因变异的病例。
1.2 研究方法
1.2.1 病史采集
回顾3个病例的病史采集表,内容包括一般情况、现病史、既往史、个人史、出生史、家族史等,通过电话回访对免疫系统、神经系统、骨骼肌肉系统、泌尿系统、视觉系统等病史进行重点问诊。
1.2.2 听力学检查
听力学检查主要包括:纯音测听(Astera headphone)、声导抗检查(GSI Tympstar)、言语识别率(解放军总医院自主研发)、畸变耳声发射(MADSEN Capella 2)、听性脑干反应潜伏期(HIS 6376),以上仪器均定时按国际标准进行校准。依据世界卫生组织(WHO)1997年制定的标准进行听力损失分级标准,按500、1000、2000、4000Hz四个频率的平均听阈进行分级:正常:≤25 dB HL;轻度:26~40 dB HL;中度:41~60 dB HL;重度:61~80 dB HL;极重度:≥81 dB HL。
1.2.3 其他辅助检查
病例1辅助检查包括血常规、尿常规、血生化、血沉(ESR)、C反应蛋白(CRP)、自身免疫抗体、脑脊液检查、眼底照相、角膜地形图、光学相干断层成像等;病例2辅助检查包括血常规、血生化、ESR、CRP、凝血四项、自身免疫抗体、血清白介素测定、颅脑CT、颅脑MRI等;病例3仅接受了血清白介素测定检查。
1.2.4 遗传学检测
签署知情同意书后抽取病例1,及父母、胞妹4人,病例2及其父母3人,病例3及其父母3人,共10人的外周静脉血5~10ml,提取DNA样本,采用Illumina HiSeq2500平台,对所有样本进行全外显子组测序(whole exome sequencing,WES)。目标区域平均测序深度108X,其中目标序列的96%深度达20X以上。对病例3家系其他成员(祖父母、叔姑等4人)进行候选基因变异位点的Sanger验证。
2 结果
2.1 一般资料
病例1,女,9岁,双耳渐进性听力下降6年。既往史:出生后半年开始患儿反复出现夜间发热伴红色风团样皮疹,其后听力及视力逐渐下降,头痛、发热、皮疹、眩晕等症状反复发作,因分泌性中耳炎曾于外院行鼓膜切开置管术。言语发育正常,出生史无异常,无家族遗传病史,有一胞妹无听力及其他系统异常。治疗:确诊后长期于外院口服激素、曲尼司特、沙利度胺等药物治疗,病情反复。
病例2,男,32岁,双耳渐进性听力下降6年余、伴高调耳鸣。无眩晕等。有反复口腔溃疡、荨麻疹、湿疹、结膜炎病史,无反复发作的发热等病史,否认关节病、肌无力等病史。否认耳毒性药物及噪声接触史。否认家族遗传病史,父母听力均正常。患者听力下降较快,于2019年开始佩戴助听器。治疗:按突发性耳聋收入我科治疗2个疗程,给予激素、鼠神经生长因子、甲钴胺、银杏叶提取物、前列地尔等药物,效果不明显。
病例3,男,24岁,双耳渐进性听力下降4年。否认眩晕、口腔溃疡、荨麻疹、湿疹、关节病、肌无力等病史。有听力下降家族遗传史,祖父、父亲均为20岁左右开始双耳渐进性听力下降,因个人原因其父亲及祖父未行听力学检查。治疗:于外院门诊按突发性耳聋间断给予甲钴胺、胞磷胆碱、单唾液酸四己糖神经节苷等药物治疗,效果不明显。
2.2 听力学检查结果及变化
病例1首诊时为双耳混合性听力损失,46个月后低中频骨导听力明显提高,左耳骨导平均阈值为23.75dB HL,右耳骨导平均阈值为21.25dB HL,转变为双侧传导性耳聋。病例2和病例3为双耳渐进性高频下降型SNHL。首诊时病例2和病例3听力损失程度分别为中度和轻度。病例2在56个月内左耳平均听阈升高11.25dB,右耳平均听阈升高21.25dB,听阈变化速率左耳2.25dB/year,右耳4.25dB/year,听力受损频率从高频累及全频,言语识别率左耳下降16%,右耳下降24%;病例3在52个月内左耳平均听阈升高28.75dB,右耳平均听阈升高15dB,听阈变化速率左耳5.75dB/year,右耳3dB/year,高频区域听力下降明显(图1)。
2.3 其他辅助检查结果
病例1有白细胞计数、CRP的反复升高,脑脊液检查提示高颅压,眼科检查提示视神经萎缩,其余辅助检查均正常。病例2视力检查、血常规、尿常规、血生化、凝血四项、红细胞沉降率、自身抗体、白介素测定、内听道水成像等指标均正常。病例3白介素测定正常。
2.4 遗传学检测结果
WES结果显示:病例1携带NLRP3(NM_001243133.1):c.907G>A(p.D303N),WES 未 发现其父母及胞妹携带该变异。病例2携带NLRP3(NM_001243133.1):c.2753G>A(p.R918Q),在其父母DNA样本WES结果中未发现该变异。病例3携带NLRP3(NM_001243133.1):c.1562T>C(p.M521T),在其父亲DNA样本WES结果中发现该变异,母亲未发现该变异,通过Sanger测序在其祖父DNA样本中发现该变异,在其祖母、叔姑3人DNA样本中未发现该变异(图2)。
2.5 突变致病性分析
p.D303N、p.R918Q是各自病例家庭中的新发变异,且均为已报道的致病变异[6,7]。病例3所携带变异p.M521T既往未见报道,根据ACMG指南,该变异被判断为疑似致病变异(PM1+PM2+PP1-moderate+PP2):PM1:位于热点突变区域,ESP数据库、千人数据库、EXAC数据库中正常对照人群中未发现的变异;PM2:在ESP6500、千人、EXAC、GnomAD等数据库中人群频率均为0;PP1-moderate:突变与疾病在家系中共分离(在家系多个患者中检测到此变异,升级为moderate);PP2:对某个基因来说,如果这个基因的错义变异是造成某种疾病的原因,并且这个基因中良性变异所占的比例很小,在这样的基因中所发现的新的错义变异。

图1 NLRP3相关听力损失病例首诊与复诊听力图Fig.1 Audiogram of first and second visit of NLRP3 related hearing loss cases

图2 病例家系图谱Fig.2 Pedigrees of cases in this study
3 讨论
NLRP3基因变异引起的CAPS是罕见的遗传性疾病,发病率在美国为1/100万,在德国为3.43/100万,在法国为1/36万,不同性别与种族不存在差异[8,9]。CAPS包括三种亚型:症状最轻的家族性寒冷性自身炎症综合征(familial cold autoinflammatory syndrome,FCAS),表现为间歇性寒冷诱导的发热、荨麻疹样皮疹、关节痛和结膜炎;中间表型Muckle-Wells 综 合 征 (Muckle-Wells syndrome,MWS),表现为皮疹、淀粉样变性和高频受累的SNHL,占青少年CAPS的三分之二[10];症状最严重的新生儿起病多系统炎症疾病(neonatal-onset multisystem in flammatory disease,NOMID),又被称为慢性婴儿神经系统皮肤和关节综合征(chronic infantile neurologic,cutaneous and articular syndrome,CINCA),症状包括婴儿起病的进行性双侧SNHL、慢性无菌性脑膜炎、周期性发热、头痛、癫痫、结膜炎、葡萄膜炎和/或视神经萎缩。本研究中病例1起病于新生儿期,临床表现严重,多系统受累,反复发热、皮疹、头痛,渐进性听力损失,视神经萎缩等,根据临床表现、辅助检查和WES结果,诊断为CINCA。该病例长期伴有分泌性中耳炎,纯音测听提示混合性听力损失,类似病例如首诊于耳鼻喉科,专科医师如缺乏对CAPS的足够认识,可能出现漏诊误诊。2017年Nakanishi等[6]鉴定NLRP3变异可以引起非综合征型常染色体显性遗传性耳聋DFNA34。病例2和病例3均发病于20岁以后,临床表现单一,双耳听力损失以高频听阈为主,逐渐累及低中频,与文献[6]报道相符,无明显全身炎性反应,无其他系统受累,结合WES结果,诊断为NLRP3相关的非综合征型耳聋。这2例病例听力阈值变化速率(病例2:左2.25dB/year,右4.25dB/year;病例3:左 5.75dB/year,右 3dB/year)高于文献[6]报道的 0.9~1.5 dB/year,可能与环境等其他因素有关。
慢性耳蜗炎症和Corti器的变性坏死是可能NLRP3变异相关耳聋的组织学病因,听力损失较重的情况下也罕见前庭功能的损伤,据此,有研究[4]推测病变可能位于螺旋韧带和血管纹病变;CAPS患者钆造影MRI成像呈现耳蜗基底部异常强化且IL-1抑制剂治疗后强化减轻[5,6,11],印证了耳蜗慢性炎症导致听力损失的假说。病例1炎性指标C反应蛋白、纤维蛋白原、白细胞计数等指标随发热反复升高,提示活跃的全身炎性反应,下一步需要补充血清白介素检测和钆造影MRI成像来更好地评估全身和局部的炎性反应。病例2和病例3炎性反应指标和血清白介素检测均正常,可完善钆造影MRI成像检查来评估内耳炎性反应情况。与病例1相比,病例2和病例3为何只有内耳局部受累而没有全身炎性反应?仍然需要更深入的研究。
迄今为止(2020年2月7日)共有230个NLRP3基因序列变异被发现,其中19个为CAPS相关致病变异,79个为CAPS相关的疑似致病变异,1个为DFNA34相关的致病变异(https://infevers.umai-montpellier.fr/web/search.php?n=4),这些变异在NLRP3基因示意图上所对应的位置及表型汇总详见图3。病例1和病例3所携带的变异均位于3号外显子,病例2所携带的变异位于7号外显子,不同变异位点引起的表型异质性是NLRP3相关疾病的研究热点:①不同变异位点造成耳聋的风险不同,如p.E311K、p.T348M变异携带者发生听损的风险比p.V198M携带者高[4];②不同变异位点造成听力损失程度不同[12],如p.T348M导致的听力损失较重,p.A439V导致听力损失程度相对较轻;③不同变异位点引起综合征累及的器官数量也不同,如Caifeng Li等[13]报道,在15例出生2天至6岁的中国CAPS患儿中p.D305N携带者表现出多脏器受累的严重表型,而p.F311S携带者则没有重要脏器的损伤;④不同变异位点导致外显率不同,如一些p.V198M,p.Q703K和p.R488K携带者不发病或表现为轻微的迟发病变,显示出较低的外显率[14,15]。不同位点引起不同表型的具体机制仍然有待研究。
NLRP3基因的新发变异在CINCA患者中较为多见[16,17],尚未在非综合征型遗传性耳聋患者中发现。本研究中病例1和病例2所携带的变异均为新发变异,这提示我们:对无家族遗传史的双侧SNHL患者,特别是出现全身炎性反应表现的双侧耳聋患者,应重视对NLRP3基因变异的筛查。病例3携带变异c.1562T>C,符合家系内常染色体显性遗传模式,Sanger测序提示在家系其他成员中共分离,根据ACMG指南可得到3个中等强度证据,1个支持证据,判定为疑似致病变异,仍需要进一步功能研究,以获得更充分的致病性证据。
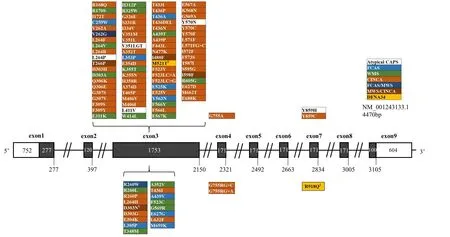
图3 已知变异在NLRP3基因上的分布图Fig.3 Distribution of known NLRP3 variations
回顾文献笔者发现:IL-1生成增多是CAPS和DFNA34共同病因,抗IL-1治疗就成为了这类疾病治疗的主要方案[18]。目前在欧美已批准上市用于临床治疗CAPS的药物包括canakinumab(IL-1β单克隆抗体),anakinra(IL-1α和IL-1β受体拮抗剂),rilonacept(IL-1α和IL-1β诱导性受体),以上3种药物对CAPS系统症状和全身炎性反应都有着良好的缓解率,可以维持听力稳定。目前上述药物在我国尚未批准上市,但国外已对3种药物进行了疗效、不良反应等的临床研究,为3种药物的选择提供了参考。笔者从患者年龄、经济承受能力、是否有中枢神经系统的损害、对药物注射的依从性等方面对3种药物的特点归纳总结详见表1。可以肯定的是对NLRP3相关性疾病的患者应该早期干预以延缓听力损失和预防听力器官损伤。canakinumab作为一种长效制剂,患者只需要每8周接受1次皮下注射,依从性更好,有研究[12]显示其对于炎性症状的总体缓解率高于anakinra(93%>75%);而anakinra体内半衰期只有4~6小时,需要每天注射才能达到控制炎性症状的作用,但其对控制中枢神经系统的炎症效果较好[19],适用年龄更小(年龄≥8月,体重≥10kg),缓解听力损失效果更确切[11,20],年花费较少。rilonacept注射频率介于上述两者之间,对听力的作用不确切,使用限制较大(>12岁)。3种药物对CAPS都有着较轻的副作用:anakinra和rilonacept主要是每日皮下注射引起的局部皮肤反应和轻度呼吸道感染[20,21];canakinumab主要是眩晕,泌尿、呼吸、消化系统的轻度感染,头痛等,少数患者因为上述不良反应住院治疗[22];而研究较多的anakinra,一些病例[20]连续使用5年后仍然没有见到严重的不良反应。抗IL-1治疗对CAPS患者系统症状和全身炎性反应都有着良好的缓解率。症状日记得分、外周血(WBC)、血小板(PLT)、血清淀粉酶(SAA)、C反应蛋白(CRP)、血沉(ESR)、听力测试、视力及眼底检查、膝关节CT、内耳钆造影磁共振、脑脊液(WBC、白蛋白、脑脊液压力)、内毒素刺激体外培养的外周血单核细胞分泌IL-1β水平等均可以作为病情评估指标[6,11,20,22]。
NLRP3基因相关的听力损失治疗效果报道不多,有学者[4]认为这种慢性炎症对听力的损伤在早期是可逆的,如在本研究中,病例1首诊时呈混合性听力损失,其后长期使用激素等控制全身炎性反应,46个月后复诊低中频骨导听力恢复正常,存在感音神经性听力损失逆转的可能,笔者推测在CINCA患者中通过药物控制全身炎性反应对听力的维持甚至逆转有着积极的作用。前瞻性队列研究[6,11,20]结果显示抗IL-1治疗后,患者听力提高或维持稳定,但只有少数病例听力可以恢复到正常,文献曾经报道过2名MWS成人患者[23,24],2名MWS患儿[25,26],和1名NOMID/CINCA青少年[27]达到了听力学痊愈;对DFNA34型耳聋家系皮下注射anakinra3个月后听力维持稳定[6],但对听力长期的保护甚至逆转作用仍需要长期观察来证明。总体而言,听力损失恢复程度与年龄相关,年龄较小听力损失恢复更明显[6,28,29];不同的NLRP3变异位点也可能影响抗IL-1的疗效:如Kuemmerle-Deschner等[12]对携带3种不同变异的MWS综合征患者进行抗IL-1治疗后听力恢复情况的比较,发现携带p.E311K突变的MWS患者听力提高程度较p.V198M或p.T348M携带者更为明显。

表1 抗-IL-1药物特点汇总表Table 1 Anti-il-1 drug characteristics
总之,本研究发现NLRP3基因变异可引起不同类型的双侧迟发型听力损失,部分病例伴有全身炎症反应等特殊的临床表现,听力下降速度快,不同位点变异表型异质性大,新发变异多见,增加了遗传学诊断的难度,既往文献显示抗IL-1治疗是有效的药物治疗方案。NLRP3相关性耳聋应该得到耳鼻喉科医师的重视。笔者推测该基因变异也可能是部分双侧突发性耳聋、梅尼埃、自身免疫性内耳病等内耳疾病的潜在致病基础,我们应借助新一代测序技术,在这类不明原因的双侧耳聋患者中开展NLRP3基因的筛查工作,并积极推动抗IL-1药物在我国的上市和临床应用。
