美国白蛾性信息素的合成研究
2020-04-24陈雪珂陈修华蒋国飞李新生
陈雪珂,陈修华,蒋国飞,李新生
(1.浙江师范大学化学与生命科学学院,浙江 金华321004;2.浙江海森药业股份有限公司,浙江 金华322104)
美国白蛾又名美国灯蛾,属鳞翅目灯蛾科,原产于北美洲,主要分布于美国和加拿大南部。该虫是世界性检疫害虫,严重影响林木生长,甚至侵入农田,危害农作物,造成减产减收,甚至绝产。1979年该虫从中朝边境传入我国,并迅速发展成为七大森林害虫之一,所以急需对其进行预防和控制[1]。
目前所采用的防治办法有:加强检疫,防止美国白蛾由疫区传入;白蛾网幕期,人工剪除网幕,就地销毁;白蛾化蛹时,人工挖蛹;各代成虫期,利用白蛾成虫趋光性,悬挂杀虫灯诱杀;草把诱集;选择高效低毒的仿生生物杀虫制剂高压喷雾防治;利用其天敌周氏啮小蜂防治白蛾[2];设置性信息素诱捕器,诱杀白蛾雄蛾。
利用美国白蛾性诱剂或环保型昆虫趋性诱杀器诱杀成虫。在成虫发生期,把诱芯放入诱捕器内,将诱捕器挂设在林间,直接诱杀雄成虫,阻断害虫交尾,降低繁殖率,达到消灭害虫的目的。采用人工合成的美国白蛾性信息素具有高度的专一性和很强的引诱力,能够非常高效地诱捕美国白蛾,达到检测和大量诱杀美国白蛾种群的目的。
欧美及日本学者于上世纪60年代末开始对该虫的性信息素生物学、化学结构鉴定、人工合成及应用等方面进行研究,但由于分析技术所限,至1989年[3-4]才彻底鉴定出美国白蛾性信息素的5 种全息组分。本文着重介绍以下两个组分的合成研究。
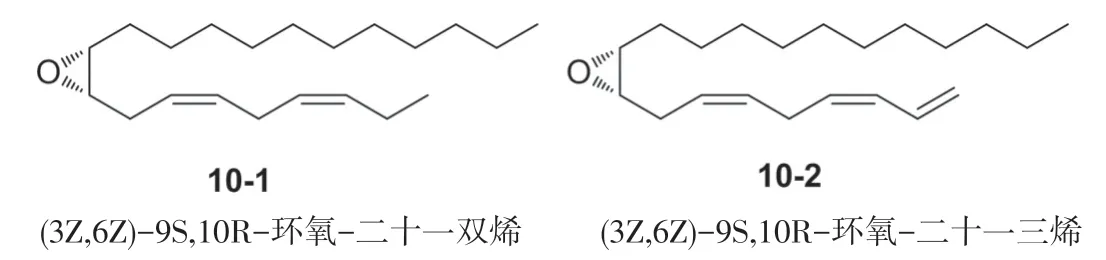
图1 目标化合物的结构及名称
鉴于美国白蛾性信息素的研究重要性,自发现之后,各国科研人员均对此进行了合成研究,1986年日本学者Mori 和Takashi[5]以1-溴-2-戊炔为原料与丙炔醇两次偶联得到脂肪三炔3,3催化加氢可得(2Z,5Z,8Z)-十一碳三烯-1-醇。经Sharpless 不对称合成环氧醇,醇羟基磺酰化后与癸烷基铜锂作用得到,合成总产率可达7%,见图2。
2002 年,倪静[12]以1,4-丁炔二醇为起始原料,由三烯醇脱水得四烯时,直接由醇脱水产生端基双键而不影响其他顺式双键,四烯醇进行Sharpless 不对称环氧化,与对甲苯磺酰氯反应成苯磺酸酯后经Witting 反应得到,图见3。
在参考2005年黄培强课题组[13]报道的合成方法下,本文以炔丙醇为起始原料,经过偶联、氢化、环氧化、溴代等反应合成化合物9,再通过不同的方法分别得到脱溴产物。其合成路线如图4所示。
1 实验部分
1.1 仪器与试剂
Bruker Avance 400 型核磁共振波谱仪(CDCl3为溶剂,TMS 为基准物);产品的纯度、粗产物中各组分的含量和反应跟踪使用气相色谱仪(Agilent 7890B);正相高效液相色谱HPLC(柱Chiralpak AD-H 4.6×250 mm;正己烷/异丙醇=95∶5;流速1.0 mL/min)。
1.2 实验方法
1.2.1 2-十四碳炔-1-醇的合成
称取炔丙醇(112.14 g,2 mol),加入300 mL 二氯甲烷稀释,加入p-TsOH·H2O(3 g),置于-10 ℃低温槽冷却降温,永恒滴液漏斗滴加3,4-二氢-2H-吡喃(168.24 g,2.2 mol),滴加结束后,自然升温反应过夜,TLC检测反应完全,加水淬灭反应,萃取,干燥浓缩,减压蒸馏收集馏分,产率92%。
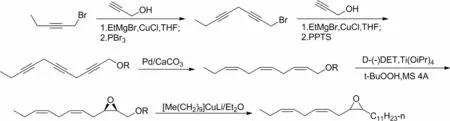
图2 Mori和Takashi的合成路线
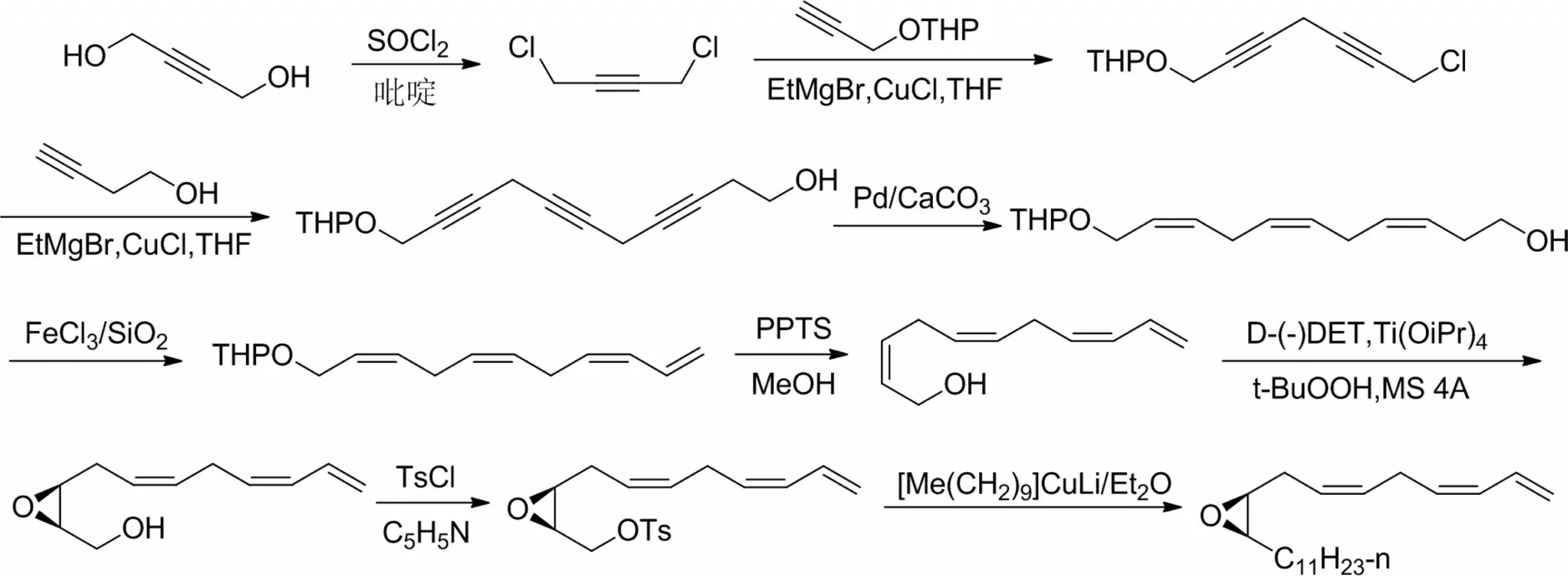
图3 倪静的合成路线
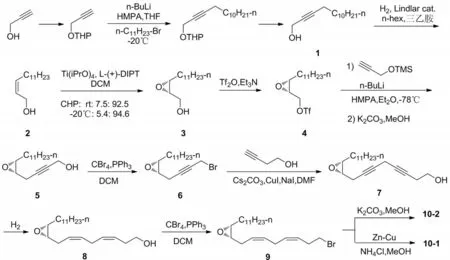
图4 合成路线
在2 L 三颈烧瓶中加入2-(2-炔丙基氧)四氢吡喃(168.2 g,1.2 mol),500 mL钠丝干燥的THF,N2保护,置于-78℃低温槽搅拌冷却,加入n-BuLi(500 mL 2.4 M,1.2 mol),反应1 h 后,加入HMPA(450 mL),1-溴十一烷(235.21 g,1 mol),自然升温反应过夜,TLC检测反应已结束,停止反应。加入NH4Cl 饱和溶液淬灭反应,旋蒸去多余的THF,乙醚萃取,干燥浓缩。
将粗产品溶于200 mL 甲醇,加入p-TsOH·H2O(5 g),加热回流5 h,TLC检测追踪反应进程,反应完成后,旋蒸除掉甲醇,乙酸乙酯萃取,盐水洗,干燥浓缩后用石油醚结晶,得到白色固体;滤液浓缩过柱(洗脱剂:V(乙酸乙酯)/V(石油醚)=1/10)。两步产率为85.1%。
1.2.2 (2Z)-2-十四碳烯-1-醇的合成
Ni(OAc)2·4H2O(74.65 g,300 mmol),加入300 mL 95%的乙醇溶液,搅拌部分溶解,置于低温浴中,加入NaBH4(13.62 g,350 mmol),待反应稳定后移至室温反应过夜,得到p-2型Ni2B。
直接向催化剂体系中加入150 g 2-十四碳炔-1-醇,40 mL乙二胺,吸氢体积足量时停止反应,核磁氢谱确定氢化程度,布氏漏斗垫硅胶抽滤,重复三次可得纯净液体,滤液干燥浓缩,得到黄色液体,不再提纯,直接进行下一步反应。
1.2.3 (2S,3R)-2,3-环氧-3-十一烷基-1-醇的合成
在2 L 三口瓶中加入CaH23 g,DCM 300 mL,N2保护置于-35 ℃低温槽,用恒压滴液漏斗逐滴加入Ti(i-PrO)4(99.48 g,350 mmol),L-(+)-DIPT(81.98 g,350 mmol),低温反应30 min 后逐滴加入(2Z)-2-十四碳烯-1-醇(106 g,500 mmol),继续反应30 min 后逐滴加入过氧化氢异丙苯(209.26 g,1.1 mol),低温反应48 h,点板,气相均显示无原料,加入10%酒石酸水溶液500 mL,继续搅拌30 min,抽滤除去固体不溶物,滤液萃取,无水Na2SO4干燥浓缩,冷却,结晶,抽滤后固体用石油醚洗,得到白色固体,滤液浓缩后通过减压蒸馏除去杂质,残余液过柱(洗脱剂:V(乙酸乙酯)/V(石油醚)=1/4),得到白色固体,总产率80.3%。
1.2.4 (5S,6R)-5,6-环氧-6-十一烷基-2-炔-1-醇的合成
称取(2S,3R)-2,3-环氧-3-十一烷基-1-醇(3 g,13.13 mmol)加入350 mL DCM溶解,氮气氛下置于-78℃低温冷却槽中,加入6 eq三乙胺、3 eq三氟甲磺酸酐,低温反应2 h,加入饱和NH4Cl 水溶液淬灭反应,萃取分液,盐水洗,有机层用无水硫酸钠干燥,浓缩通过柱色谱法快速分离(洗脱剂:V(乙酸乙酯)/V(石油醚)=1/100),得到微黄色油状液体。
反应管中加入丙炔氧基三甲基硅烷、Et2O,氮气氛下置于-78℃冷肼中冷却,加入1.1 eq n-BuLi(2.5 M),反应30 min 后加入HMPA,继续反应2 h,加入0.5 g K2CO3、20 mL MeOH,室温搅拌1 h,TLC显示反应完全,旋蒸除去溶剂,乙醚萃取,干燥浓缩过柱(洗脱剂:V(乙酸乙酯)/V(石油醚)=1/10),得白色固体,两步产率34.6%。
1.2.5 1-溴-(5S,6R)-5,6-环氧-6-十一烷基-2-己炔的合成
在冰浴下,将四溴化碳(575 mg,1.74 mmol)和三苯基膦(456 mg,1.74 mmol)加入到(5S,6R)-14(420 mg,1.58 mmol)的DCM 溶液(20 mL)中。在0℃下搅拌3 h后,将混合物浓缩除去DCM 后,加入石油醚(15 mL)稀释,有白色固体析出,抽滤除去固体,并将滤液真空浓缩。通过硅胶快速柱色谱法(Rf为0.72,洗脱液:V(乙酸乙酯)/V(石油醚)=1/40)纯化残余物,得到化合物(410 mg,78.8%),为白色固体。
1.2.6 (9S,10R)-9,10-环氧-10-十一烷基-3,6-二炔-1-醇的合成
向搅拌的细磨和无水Cs2CO3(480 g,1.47 mmol),NaI(221 mg,1.47 mmol),CuI(280mg,1.47 mmol)的无水DMF(5 mL)悬浮液中依次加入化合物6(410 mg,1.25 mmol)的无水DMF(5 mL)溶液和3-丁炔-1-醇(92 mg,1.31 mmol)的无水DMF(5 mL)溶液,在氮气氛下,将该悬浮液在室温下搅拌36 h,然后用NH4Cl 水溶液淬灭。用乙醚(2.5 mL×8)萃取混合物,将合并的有机层用无水Na2SO4干燥,过滤并真空浓缩。通过硅胶快速柱色谱法(Rf为0.25,洗脱液:V(乙酸乙酯)/V(石油醚)=1/4)纯化残余物,得到(9S,10R)-7(336 mg,84.5%),为白色固体。
1.2.7 (9S,10R)-9,10-环氧-10-十一烷基-3,6-辛二烯-1-醇的合成
P-2型Ni2B 1 g,加入3 mL乙二胺,20 mL无水乙醇作溶剂,在氢气氛下室温搅拌30 min,加入化合物7(1 g),继续反应吸氢足量后,TLC检测反应,核磁氢谱确定氢化程度,在滤纸上垫层硅胶滤除催化剂,干燥浓缩后不再纯化,直接投入下一步反应。
1.2.8 (9S,10R)-1-溴-9,10-环氧-(3Z,6Z)-二十一碳-3,6-二烯的合成
在冰浴下,将四溴化碳(270 mg,0.81 mmol)和三苯基膦(212 mg,0.81 mmol)加入到化合物8(194 mg,0.73 mmol)的DCM溶液(7 mL)中,在0℃下搅拌3 h后,将混合物浓缩除去DCM,加入石油醚(15 mL)稀释,有白色固体析出,抽滤并将滤液真空浓缩。通过硅胶快速柱色谱法(Rf为0.7,洗脱液:V(乙酸乙酯)/V(石油醚)=1/40)纯化残余物,得到化合物(202 mg,71.8%),为无色油状物。
1.2.9 (3Z,6Z)-9S,10R-环氧-二十一双烯的合成
向搅拌的CuSO4(400 mg)水溶液(20 mL)中加入锌粉(3.0 g),搅拌反应20 min后停止搅拌,待固体沉淀下来后轻轻倒出上清液,依次用水和MeOH 洗涤残余物。加入NH4Cl 饱和的MeOH(15 mL)和化合物9(1.04 g,3.22 mmol),将混合物搅拌回流60 min,通过硅胶过滤,乙酸乙酯洗涤滤饼。合并滤液,浓缩,乙酸乙酯萃取。合并的有机层用盐水洗涤,用无水Na2SO4干燥,过滤,并减压浓缩。通过硅胶快速柱色谱法纯化(Rf为0.35,洗脱液:V(乙酸乙酯)/V(石油醚)=1/50)得到化合物10-1(0.59 g,59.8%),为无色油状物。
1.2.10 (3Z,6Z)-9S,10R-环氧-二十一三烯的合成
向化合物9(0.1 g,0.26 mmol)的10 mL无水甲醇中加入无水K2CO3(0.1 g),在室温下反应48 h 后,将反应混合物过滤并真空浓缩,将残余物纯化,用硅胶快速柱色谱法(Rf为0.35,洗脱液:V(乙酸乙酯)/V(石油醚)=1/50)得到化合物10-2(56 mg,70.7%),为无色油状物。
2 结果与讨论
2.1 (2S,3R)-2,3-环氧-3-十一烷基-1-醇的合成条件的优化
N2保护下的三颈烧瓶中盛有CaH259 mg,干燥的二氯 甲 烷20 mL,50 mg 硅 胶,Ti(i-PrO)4(1.3 g,4.6 mmol),在设置温度下放置30 min,加入L-(+)-DIPT(4.6 mmol),反应30 min 后,加入(2Z)-2-十四碳烯-1-醇(0.85 g,4 mmol),过氧化氢异丙苯(9 mmol),控制温度反应2天,点板检测无原料,加入酒石酸水溶液,继续搅拌30 min,抽滤除去固体不溶物,滤液萃取,无水Na2SO4干燥浓缩,柱层析(洗脱剂:V(乙酸乙酯)/V(石油醚)=1/3)得到白色固体。
在Sharpless 不对称环氧化一步中,探究了室温与-35℃两种温度控制条件,同时也探究了Ti(i-PrO)4和L-(+)-DIPT的量的反应条件。
以1.15 eq 的Ti(i-PrO)4和L-(+)-DIPT 进行反应,结束后,通过HPLC 分析相应的3,5-二硝基苯甲酰酯(柱Chiralpak AD-H 4.6×250 mm;正己烷/异丙醇=95∶5;流速1.0 mL/min)测定环氧化物(2S,3R)-12 的对映体过量:室温的产物,(2R,3S)-酯:11.93 min(7.5%),(2S,3R)-酯:14.16 min(92.5%),ee 85.0%;-35℃的产品,(2R,3S)-酯:11.93 min(5.4%),(2S,3R)-酯:14.16 min(94.6%),ee 89.2%。因此,本步骤采用-35℃的条件进行反应。
在-35℃反应,使Ti(i-PrO)4和L-(+)-DIPT 两者均分别为0.2 eq,0.5 eq,0.7 eq时,发现在0.7 eq 时就可得到90%对映体过剩的产物,因此,后续本步骤采用-35℃,0.7 eq Ti(i-PrO)4和L-(+)-DIPT的条件进行反应。
因为优化反应均为小试,所以加料时速度较快,未控制加料速度,而在放大反应时,采取了逐滴加入的方法,得到了更为优秀的对映体过量的产物,可达97%。
2.2 产物的结构表征
(1)化合物1:2-十四碳炔-1-醇,白色固体,产率85.1%。1H NMR(400 MHz,CDCl3)δ 4.25(s,2H),2.21(ddd,J=7.1,5.0,2.1 Hz,2H),1.55-1.45(m,2H),1.41-1.33(m,2H),1.26(s,14H),0.88(t,J=6.8 Hz,3H)。
(2)化合物2:(2Z)-2-十四碳烯-1-醇,无色液体,产率97.2%,Z/E>99。1H NMR(400 MHz,CDCl3)δ 4.25(t,J=2.1 Hz,2H),4.20(d,J=6.1 Hz,1H),2.26-2.15(m,2H),1.77(s,2H),1.56-1.43(m,2H),1.42-1.16(m,17H),0.88(t,J=6.8 Hz,3H)。
(3)化合物3:(2S,3R)-2,3-环氧-3-十一烷基-1-醇,白色固体,结晶产率72.8%。通过HPLC分析相应的3,5-二硝基苯甲酰酯(柱Chiralpak AD-H 4.6×250 mm;正己烷/异丙醇=95∶5;流速1.0 mL/min)测定环氧化物(2S,3R)-12 的对映体过量:重结晶前的产物,(2R,3S)-酯:11.93 min(1.3%),(2S,3R)-酯:14.16 min(98.7%),ee 97.4%;一次重结晶之后的产品,(2R,3S)-酯:11.93 min(0.01%),(2S,3R)-酯:14.16 min(99.9%),ee 99.8%。1H NMR(400 MHz,CDCl3)δ 3.86(d,J=11.6 Hz,1H),3.68(dd,J=12.0,7.1 Hz,1H),3.16(dt,J=7.0,4.2 Hz,1H),3.08-2.99(m,1H),1.68(d,J=30.6 Hz,4H),1.28(d,J=15.3 Hz,17H),0.87(d,J=7.0 Hz,3H)。
(4)化合物5:(5S,6R)-5,6-环氧-6-十一烷基-2-炔-1-醇,白色固体,两步产率34.6%。1H NMR(400 MHz,CDCl3)δ 4.27(s,2H), 3.20- 3.08(m, 1H),3.06-2.92(m,1H),2.90-2.81(m,1H),2.60(dddd,J=14.8,5.8,4.5,2.2 Hz,1H),2.35(ddt,J=17.2,6.8,2.2 Hz,1H),1.55-1.16(m,26H),0.88(t,J=6.8 Hz,4H)。
(5)化合物7:(9S,10R)-9,10-环氧-10-十一烷基-3,6-二炔-1-醇,白色固体,产率84.5%。1H NMR(400 MHz,CDCl3)δ 4.05(t,J=6.1 Hz,2H),3.53-3.42(m,3H),3.29(d,J=4.2 Hz,1H),2.94-2.83(m,1H),2.82-2.74(m,2H),2.62(ddt,J=17.1,6.9,2.4 Hz,1H),2.10(s,1H),1.92-1.50(m,21H),1.22(t,J=6.8 Hz,3H);MS(ESI)m/z 341.3(M+Na+,100%)。
(6)化合物8:(9S,10R)-9,10-环氧-10-十一烷基-3,6-辛二烯-1-醇,微黄色固体,产率92%。1H NMR(400 MHz,CDCl3)δ 4.05(t,J=6.1 Hz,2H),3.53-3.42(m,3H),3.29(d,J=4.2 Hz,1H),2.94-2.83(m,1H),2.82-2.74(m,2H),2.62(ddt,J=17.1,6.9,2.4 Hz,1H),2.10(s,1H),1.92-1.50(m,21H),1.22(t,J=6.8 Hz,3H)。
(7)化合物10-1:(3Z,6Z)-9S,10R-环氧-二十一双烯,无色油状,产率59.8%。1H NMR(400 MHz,CDCl3)δ 5.57-5.25(m,4H),3.01-2.87(m,2H),2.80(t,J=7.0 Hz,2H),2.40(dt,J=12.5,6.1 Hz,1H),2.28-2.14(m,1H),2.13-1.99(m,2H),1.61-1.19(m,20H),0.97(dd,J=9.6,5.5 Hz,2H),0.88(dd,J=8.6,5.0 Hz,3H);MS(ESI)m/z 329.2(M+Na+,100%)。
(8)化合物10-2:(3Z,6Z)-9S,10R-环氧-二十一三烯,无色油状,产率70.7%。1H NMR(400 MHz,CDCl3)δ 7.07-6.90(m,1H),6.36(s,1H),5.94-5.67(m,2H),5.59-5.43(m,1H),3.35-3.20(m,3H),2.72(s,1H),2.55(s,1H),1.99-1.52(m,20H),1.22(t,J=6.8 Hz,3H);MS(ESI)m/z 327.2(M+Na+,100%)。
3 结论
以炔丙醇为起始原料,经过偶联、氢化、环氧化等反应合成中间体化合物3,再通过偶联、溴代等得到中间体9,最终经不同的方法分别得到脱溴产物,得到高纯度的美国白蛾性信息素。本研究优化了反应条件:①使用P-2 型Ni2B,氢化烯烃得到纯顺式加成的烯烃产物;②在Sharpless 不对称环氧化一步中,可放大反应至500 mmol,仍可得到99.8%对映体过剩的产物。
