甲基黄药和丁基黄药在黄铜矿(001)和(112)面的吸附机理研究
2020-04-13范瑞华李育彪魏桢伦
范瑞华 李育彪 魏桢伦
(武汉理工大学资源与环境工程学院,湖北武汉430070)
黄铜矿(CuFeS2)是铜在自然界中最主要的赋存矿物[1],世界铜储量的70%存在于黄铜矿中[2]。黄铜矿往往与其他硫化矿致密共生,嵌布关系复杂[3],因此需要细磨才能达到单体解离。但是,在磨矿过程中,黄铜矿表面会暴露出许多不同晶面。研究表明,黄铜矿的不同晶面会发生重构并产生性质差异[4]。但是,有关黄铜矿不同晶面性质差异的报道较少。
黄铜矿通常采用浮选的方法进行富集,在浮选分离过程中,黄药是应用最为广泛的一种捕收剂[5,6],国内外有关黄药与黄铜矿的作用机理报道较多[7-10],但浮选过程中黄药在黄铜矿不同晶面上的作用机理并不明确,导致不同研究者得到的浮选结果差异性较大。因此,在浮选过程中需要考虑黄铜矿不同晶面的影响。
基于密度泛函理论的计算方法近年来被广泛应用于硫化矿的浮选当中[4,11-15],Cláudio de Oliveira等用密度泛函方法研究了黄铜矿各晶面的弛豫[4],以及水分子在黄铜矿(001)面上的吸附[13];李育彪课题组研究了一价阳离子与二价阳离子在黄铜矿(001)面上的吸附[14,15]。但是,黄铜矿(112)面也是黄铜矿的主要晶面之一[16]。因此,本文旨在通过密度泛函理论,模拟计算黄药在黄铜矿典型晶面(001)面[17]和(112)面[18]上的吸附,为研究黄铜矿不同晶面与黄药之间的作用机理提供参考。
1 计算方法
1.1 计算参数设置
采用Materials studio软件中的Castep模块进行计算,对交换关联泛函修正近似时选用广义梯度近似(GGA)下的PW91梯度,采用超软贋势(USP)对价电子和离子的相互作用势进行描述,计算中所选取的截断能为351 eV,Brillouin区的积分计算采用3×3×1的Monkhorst-Packk点网络。进行几何优化(Geometry Optimization)计算时采用BFGS算法,对黄铜矿进行优化的收敛标准按照medium标准下的相关参数进行,所有计算均采用了自旋极化。
1.2 表面能计算方法
表面能计算方法如式(1)所示[19],

式中,Esurf为表面能,J/m2;Eslab为晶面总能量,eV;Ebulk为原始单胞总能量,eV;Nslab为晶面的原子数,Nbulk为原始单胞的原子数目,A表示晶面的表面积,m2。计算得到的表面能数值越小,则表明此晶面越稳定。
1.3 吸附能计算方法
通过吸附能的数值可以确定该反应能否自发进行。吸附能计算方法如公式(2)所示[17]:
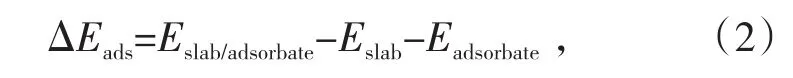
式中,ΔEads为吸附能,kcal/mol;Eslab/adsorbate为反应完成后整个体系总能量,kcal/mol;Eslab为反应前晶面总能量,kcal/mol;Eadsorbate为反应前吸附质的能量,kcal/mol。若吸附能大于零,则不能自发进行,若吸附能小于零,则可以自发进行,且吸附能越小,该反应越容易自发进行。
2 试验结果与讨论
2.1 黄铜矿(001)-M面和(112)-M面的弛豫
计算所用晶胞参数与文献[15]一致。已有研究表明[13,20],黄铜矿表面S原子表现出疏水性,Fe、Cu原子表现出亲水性。因此,本文仅对黄铜矿的金属表面,即(001)-M面和(112)-M面进行研究。从黄铜矿单胞中切割出(001)-M面和(112)-M面,然后在c方向构建厚度为15 Å的真空层,以此避免层间相互作用,最后使用Castep模块在所选参数下对晶面进行弛豫。弛豫后的晶面结构如图1所示。通过与黄铜矿原晶胞对比,发现黄铜矿(001)-M面弛豫非常小,没有产生明显的结构重组现象。黄铜矿(112)-M面表面的Cu原子在c方向上产生了位移,向晶胞内移动,使结构趋于稳定。根据计算结果得到黄铜矿(001)-M面和(112)-M面的表面能分别为1.566 J/m和1.018 J/m。由此可知,黄铜矿(001)-M面比(112)-M面稳定性差,更易与浮选药剂发生作用。

2.2 黄药在黄铜矿(001)-M面和(112)-M面的吸附
选取丁基黄原酸钠和甲基黄原酸钠作为捕收剂[21,22],考察其在黄铜矿(001)-M面和(112)-M面的吸附。图2所示为结构优化后的甲基黄原酸钠与丁基黄原酸钠的Mulliken电荷。

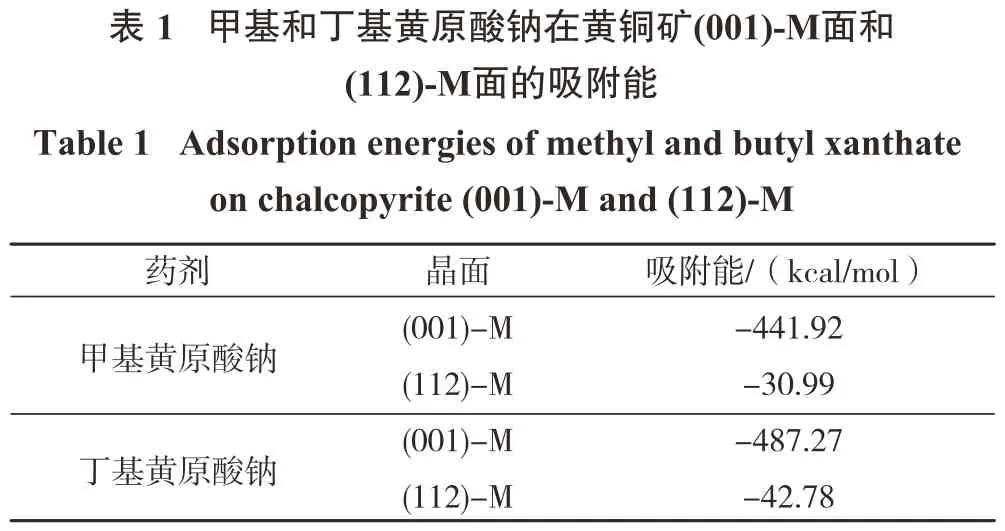
将优化后的甲基黄原酸钠和丁基黄原酸钠分别置入黄铜矿表面的2×2超胞进行弛豫,计算得到甲基黄原酸根离子和丁基黄原酸根离子在黄铜矿(001)-M面和(112)-M面的吸附能,如表1所示。
从表1可以看出,甲基黄原酸根离子和丁基黄原酸根离子在黄铜矿(001)-M面和(112)-M面的吸附能均小于零,说明两者都能自发且容易在黄铜矿(001)-M面和(112)-M面发生吸附。并且,丁基黄原酸根离子在两表面的吸附能均强于甲基黄原酸根离子,结合Mulliken电荷图中其所带负电荷强于甲基黄原酸根离子,说明碳链越长的黄药类捕收剂,越容易与矿物表面发生作用,这与文献[23]一致。
结合电荷密度图(图3)可知:甲基黄原酸根离子是通过C—S单键中的S原子与黄铜矿(001)-M面的Cu原子形成单键,丁基黄原酸根离子则是通过末端的2个S原子分别与(001)-M面的Cu原子和Fe原子成键;甲基黄原酸根离子与丁基黄原酸根离子均未在(112)-M面生成化学键,结合Mulliken电荷图分析可知,2种黄原酸根离子主要通过S原子与黄铜矿(112)-M面上的金属原子通过静电作用而吸附。

表2所示为黄原酸根离子在黄铜矿(001)-M吸附后的Mulliken布居值。
从表2可以看出,甲基黄原酸根离子在(001)-M面上形成S—Cu键的Mulliken布居值为0.35,而丁基黄原酸根离子在(001)-M面上形成的S—Fe键与S—Cu键的Mulliken布居值分别为0.42与0.43,均大于前者。布居值越大,则所成键共价性越强且更加稳定[24],因此,丁基黄原酸根离子在黄铜矿(001)-M面的吸附更强,成键更稳定。

3 结论
(1)黄铜矿(001)-M面弛豫后无明显变化,(112)-M面的表面Cu原子向晶胞内移动使结构趋于稳定。通过表面能的对比发现,(112)-M面比(001)-M面更稳定。
(2)甲基和丁基黄原酸钠主要通过静电吸附形式与(112)-M面作用,而在(001)-M面,甲基黄原酸钠主要通过C—S单键中的S原子与黄铜矿表面Cu原子成键,丁基黄原酸钠则主要通过末端的2个S原子分别与Cu原子和Fe原子成键。
