二甲双胍逆转低氧诱导的胰腺癌细胞铁死亡抵抗及潜在的作用机制
2020-04-11蔡晓杰王鸣高洁姜伟宋廉张礼荣龚爱华朱海涛王冬青
蔡晓杰, 王鸣, 高洁, 姜伟, 宋廉,张礼荣, 龚爱华, 朱海涛, 王冬青
(1. 江苏大学医学院, 江苏 镇江 212013; 2. 江苏大学附属医院影像科, 江苏 镇江 212001)
胰腺癌已经成为仅次于肺癌的第二大癌症致死肿瘤[1],且导致胰腺癌恶性程度极高的主要原因是化疗抵抗,而肿瘤微环境在胰腺癌治疗抵抗中发挥重要的作用。低氧是胰腺癌微环境最重要的特点之一[2]。低氧及其诱导的低氧诱导因子1α(hypoxia-inducible factor 1α,HIF-1α)参与多种肿瘤靶基因的转录调控,进而影响肿瘤细胞的能量代谢、增殖和凋亡,是导致放化疗诱导细胞死亡的最重要抵抗因素[3-4]。因此,有效的逆转低氧在肿瘤细胞死亡中的作用具有重要的意义。
铁死亡是一种新型死亡模式,其本质是细胞内脂质氧化物的代谢障碍,进而在铁离子的催化下产生大量脂质过氧化物,消耗抗氧化物,破坏细胞内氧化还原平衡,触发细胞死亡[5]。已有研究表明铁死亡诱导剂联合吉西他滨可有效抑制胰腺癌的增殖,促进细胞死亡[6],但是这一联合效应却仍可以被低氧及其诱导的HIF-1α所缓解[7]。二甲双胍是调节细胞糖代谢的传统药物, 其可通过腺苷酸活化蛋白激酶-雷帕霉素靶蛋白(AMP-activated protein kinase/mammalian target of rapamycin,AMPK-mTOR)通路调控HIF-1α的表达[8],同时也有研究提示雷帕霉素靶蛋白(mammalian target of rapamycin,mTOR)途径抑制剂雷帕霉素能够促进肿瘤细胞铁死亡[9]。因此,本研究拟探讨二甲双胍能否通过抑制HIF-1α进而逆转低氧引起的肿瘤细胞铁死亡抵抗效应,为胰腺癌的化疗提供新的策略。
1 材料与方法
1.1 材料
人胰腺癌Patu8988细胞株 (中国科学院上海细胞研究所);DMEM高糖培养基,PBS(美国Hyclone公司);澳洲胎牛血清(美国Gibco公司);二甲双胍、Erastin、铁死亡抑制剂ferrostatin-1、凋亡抑制剂ZVAD-FMK、坏死抑制剂necrosulfonamide(美国MedChemExpress公司);兔抗人多克隆抗体HIF-1α(美国Abcam公司);兔抗人单克隆抗体β-微管蛋白,兔抗人多克隆抗体p-AMPK、AMPK、p-mTOR、mTOR、谷胱甘肽过氧化物酶4(glutathione peroxidase 4, GPX4)(美国Cell 公司);羊抗兔二抗(美国Santa Cruz公司);6孔细胞培养板、96孔细胞培养板(美国Corning公司);CCK8试剂盒(北京智杰方远科技有限公司)。
1.2 方法
1.2.1 细胞培养 常氧培养:Patu8988细胞培养在含10 %胎牛血清的高糖DMEM中,置于37 ℃、5% CO2细胞培养箱中。低氧培养:将细胞置于密封的低氧培养盒中,向低氧培养盒里充入低氧混合气体(1% O2,5% CO2,94% N2,购自南大恒通气体厂),持续充入10 min然后将低氧培养盒转移到37 ℃培养箱中培养。
1.2.2 CCK-8法检测细胞活性 取对数生长期Patu8988细胞接种于96孔板,每孔5 000个细胞,每组设3个复孔;次日细胞贴壁后,经或不经药物处理,分别置于常氧和低氧培养条件下;待达到作用时间,配置CCK-8反应溶液(90 μL无血清培养基+10 μL CCK-8试剂);取出96孔板,每孔加入100 μL反应溶液,同时设立空白组(只含DMEM)进行校正;分别置于不同培养条件培养2 h;用酶标仪测定450 nm波长处各孔光密度(D)值。每组实验独立重复3次。计算细胞活性=实验组-空白组/对照组-空白组。
1.2.2.1 常氧和低氧培养不同时间细胞的活性 将细胞接种于96孔板后置于常氧和低氧培养条件,分别在0、12、24、48 h时间点取出,加入CCK-8检测各孔细胞活性。
1.2.2.2 经不同浓度的Erastin处理不同时间后细胞的活性 用培养基将Erastin浓度稀释为0、2.5、5、10、20 μmol/L,分别取100 μL与细胞共孵育;置于常氧和低氧培养条件,分别在0、12、24、48 h时间点取出,加入CCK-8检测各孔细胞活性。
1.2.2.3 经不同药物分组处理后细胞的活性 细胞与各组药物(对照组、10 μmol/L Erastin组、40 mmol/L二甲双胍组、10 μmol/L Erastin+40 mmol/L二甲双胍组、10 μmol/L Erastin+40 mmol/L二甲双胍+1 μmol/L Ferrostatin组、10 μmol/L Erastin+40 mmol/L二甲双胍+1 μmol/L ZVAD-FMK组、Erastin+40 mmol/L二甲双胍+1 μmol/L necrosulfonamide组)在低氧条件下共孵育24 h后取出,加入CCK-8检测各孔细胞活性。
1.2.2.4 经不同浓度二甲双胍处理后细胞的活性 用培养基将二甲双胍浓度稀释为0、20、40、80 mmol/L,分别取100 μL与细胞在低氧条件下共孵育24 h后取出,加入CCK-8检测各孔细胞活性。
1.2.3 蛋白质印迹法检测细胞相关蛋白的表达 收集Patu8988细胞,加入蛋白裂解液,提取总蛋白,100 ℃煮沸5 min;离心机12 000×g离心5 min,所得上清液即为蛋白样品。以每个泳道20 μL蛋白样品上样,行10 % SDS-PAGE;将蛋白转移至PVDF膜;5%脱脂牛奶室温封闭1 h;加入一抗,4 ℃孵育过夜;次日TBST洗膜3次,每次10 min;二抗37 ℃孵育1 h;TBST洗膜3次,每次10 min;ECL发光试剂显影,在凝胶成像系统上拍照并分析。一抗分别为兔抗人多克隆抗体HIF-1α,兔抗人多克隆抗体GPX4,兔抗人多克隆抗体p-AMPK, 兔抗人多克隆抗体AMPK,兔抗人多克隆抗体p-mTOR,兔抗人多克隆抗体mTOR,内参为兔抗人单克隆抗体β微管蛋白(均为1 ∶1 000);二抗为羊抗兔二抗(1 ∶10 000)。
1.3 统计学分析

2 结果
2.1 低氧抑制胰腺癌细胞活性
与常氧组相比,低氧组Patu8988细胞活性降低,且低氧培养24、48 h细胞活性明显低于常氧组(t=2.83、13.16,P<0.05)。见图1。由此说明低氧能够抑制胰腺癌细胞活性。
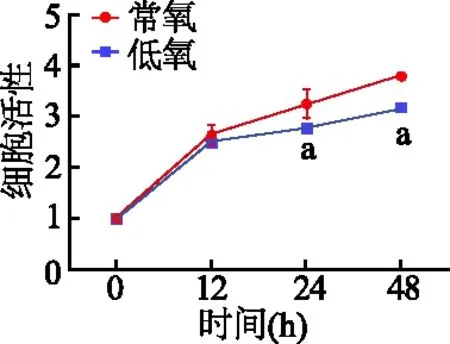
a:P<0.05,与常氧组比较
2.2 低氧使得胰腺癌细胞发生铁死亡抵抗
与对照组相比,不同浓度的铁死亡诱导剂Erastin处理24 h后,常氧或低氧培养的Patu8988细胞的细胞活性均减低。在同一Erastin浓度处理条件下,与常氧组相比,低氧组Patu8988细胞活性高于常氧组。且当Erastin浓度为10 μmol/L时,两组间细胞活性存在最显著的差异(t=3.51,P<0.05)。故选择10 μmol/L 作为Erastin后续处理浓度。见图2。

a:P<0.05,与常氧组比较
由图3可见,与对照组相比,Erastin分别处理常氧和低氧培养条件下的Patu8988细胞 12、24和48 h,Patu8988细胞的活性均减低。在同一培养时长条件下,与常氧组相比,低氧组细胞的细胞活性高于常氧组,且在24 h差异最大(t=2.46,P<0.05)。由图4可见,低氧条件下,Patu8988细胞中HIF-1α的蛋白表达水平呈一定的时间依赖性,在24 h时,蛋白表达水平最高(t=40.68,P<0.05)。由此可见,低氧条件下细胞HIF-1α表达变化与Erastin对细胞的活性变化存在一定的一致性。

a:P<0.05,与低氧组比较

a:P<0.05,与低氧0 h比较
2.3 二甲双胍增敏低氧条件下Erastin诱导的铁死亡效应
与对照组相比,低氧条件下Erastin,二甲双胍均可抑制Patu8988细胞的活性。与单独Erastin组相比,Erastin+二甲双胍抑制作用更加显著(t=58.45,P<0.01)。与联合用药组相比,外加铁死亡特异性挽救剂Ferrostatin后,细胞明显恢复活性(t=10.77,P<0.05),而外加凋亡抑制剂ZVAD-FMK组和necrosulfonamide组与联合组比,差异均无统计学意义。由此说明,联合用药是通过增强铁死亡效应来进一步抑制细胞活性。见图5。蛋白印迹显示,单独使用Erastin处理后GPX4蛋白水平降低,Erastin联合二甲双胍处理后与单独Erastin组相比进一步降低了GPX4蛋白水平(t=3.05,P<0.05),同时抑制了HIF-1α的表达(t=10.95,P<0.05)。见图6。进一步说明二甲双胍可以逆转低氧诱导的Patu8988细胞对铁死亡诱导剂Erastin的抵抗。

a:P<0.01,与未经药物处理组比较;b:P<0.05,与Erastin+二甲双胍组比较
图5各组药物作用24 h后对低氧下细胞活性的影响
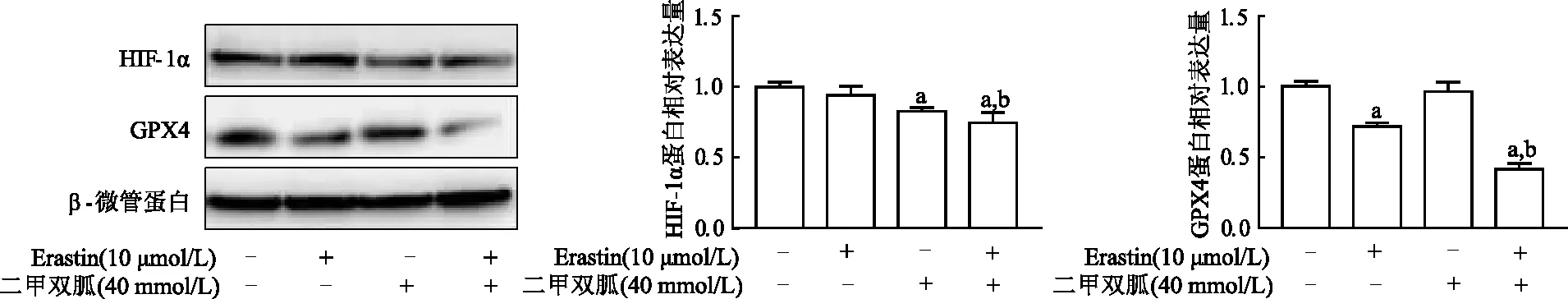
a:P<0.01,与未经药物处理组比较; b: P<0.05,与Erastin组比较
2.4 低氧条件下二甲双胍阻断HIF-1α的积聚
在低氧条件下使用不同浓度的二甲双胍作用于细胞24 h,图7可见,随二甲双胍浓度递增,细胞活性呈浓度依赖性降低。蛋白印迹结果显示,二甲双胍浓度递增促进了AMPK的磷酸化,同时抑制其下游的重要信号分子mTOR的磷酸化,进而显著降低了HIF-1α蛋白水平,见图8。从而验证了AMPK-mTOR/HIF-1α信号通路在二甲双胍抑制胰腺癌中发挥重要作用。
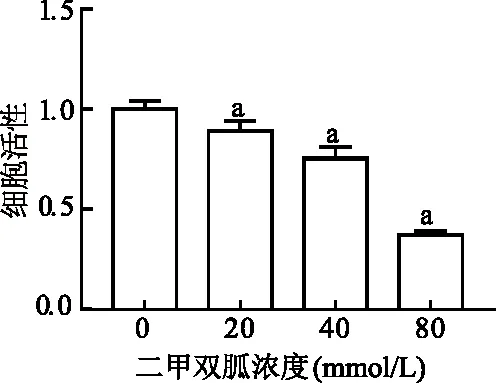
a:P<0.05,与0 mmol/L二甲双胍处理组比较

a:P<0.05;b:P<0.01,与0 mmol/L二甲双胍处理组比较
3 讨论
铁死亡是一种由铁离子催化、多不饱和脂肪酸的过度氧化而诱导的一种新型细胞程序性死亡模式[5]。脂质过氧化形成导致活性氧过度积累是诱发细胞铁死亡的关键因素[9]。GPX4是铁死亡发生的重要指标,一旦失效,会造成膜脂上活性氧自由基的积累,引发铁死亡。铁死亡概念的提出为多种实体瘤的治疗提出了新的理念[10-12]。研究发现,青蒿琥酯可诱导Kras突变的胰腺癌细胞铁死亡[13];联合荜拨明碱、植物生长调节剂CN-A 和柳氮磺胺吡啶可以有效地抑制胰腺癌的生长,主要是通过诱导铁死亡作用[14]。本研究发现,铁死亡诱导剂Erastin可以诱导人胰腺癌Patu8988细胞铁死亡,这与以往的报道一致[15]。虽然针对铁死亡的研究有效地改善了肿瘤的抑制效应,但是部分肿瘤仍具有铁死亡抵抗的特征。有研究发现,肿瘤细胞可通过Prominin2 MVB外显子铁蛋白途径抵抗铁死亡[16],也可以通过cadherin-NF2-Hippo-YAP通路负向调控铁死亡[17]。尽管针对铁死亡的抵抗机制做了深入的研究,但是绝大多数的研究都是基于细胞自身的因素。而微环境因素对肿瘤细胞铁死亡的作用机制还不是十分明确。本研究结果提示低氧可以促进胰腺癌细胞的铁死亡抵抗。
低氧是胰腺癌肿瘤微环境的最重要特征之一。低氧通过激活HIF-1α调控多种信号分子,进而影响肿瘤的放疗、化疗抵抗、转移及肿瘤血管生成等生物学行为[3-4,18]。本研究发现,低氧条件下,人胰腺癌细胞对铁死亡诱导剂Erastin抵抗性增强,呈一定的时间依赖性。且抵抗的时间依赖性与HIF-1α表达变化趋势一致,提示HIF-1α在低氧胰腺癌中起着负向调控铁死亡的作用。我们推测:细胞内积累的HIF-1α抑制低氧引起的活性氧的产生,进而抑制活性氧积累引起的铁死亡。这一效应仍需要更多的实验数据来证实。
二甲双胍抗肿瘤得到了越来越多的关注。但是当前二甲双胍使用浓度远大于临床治疗浓度[19],因此临床转化存在风险。如果能够将二甲双胍和其他抑癌药物联合使用,可以有效地降低二甲双胍的使用浓度。二甲双胍抗肿瘤机制之一是通过激活AMPK抑制mTOR从而抑制上述HIF-1α的表达[20]。我们的研究结果证实,低氧条件下在Patu8988细胞中随二甲双胍浓度递增AMPK激活,mTOR磷酸化受到抑制,HIF-1α表达也随之下调。同时,联用二甲双胍和铁死亡诱导剂Erastin与单独用Erastin组比,有效地减少了HIF-1α的聚集,降低了GPX4的表达,增强了铁死亡效应。低氧条件下二甲双胍逆转铁死亡抵抗的示意图概述见图9。
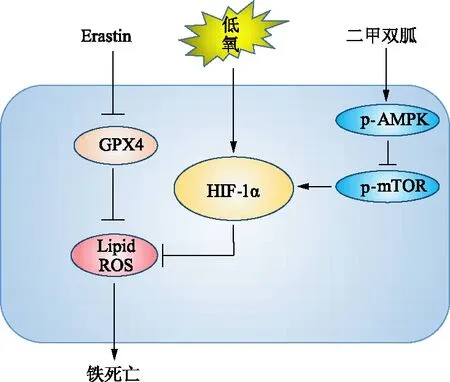
图9 低氧下二甲双胍逆转铁死亡抵抗原理示意图
然而在上述过程中二甲双胍对HIF-1α的抑制是否主要由AMPK-mTOR途径调控还有待于进一步研究。 综上所述,二甲双胍可以通过AMPK-mTOR/HIF-1α信号通路减弱低氧引起的胰腺癌细胞对铁死亡抵抗。
