基于Abraham模型估算有机物蒸发焓
2020-04-04左丽华陈六平
左丽华,陈六平
(1. 东华理工大学 化学生物与材料科学学院,江西 南昌 330013;2. 中山大学 化学学院,广东 广州 510275)
蒸发焓是物质重要的热力学性质,是化工过程计算、分析和设计的重要基础物性数据之一。随着化学工业的发展,对物质蒸发焓数据的准确度要求越来越高、数量要求越来越多。常用蒸发焓数据的获取方法主要有:量热法[1]、蒸气压法[2]、估算法。由量热法直接测出的蒸发焓数据相当可靠,但已测的数据量少,不能满足科学研究与工程设计的需要。蒸气压法则是利用Clausius-Clapeyron方程由蒸气压数据求解,数据的可靠性比量热法的可靠性低很多。蒸发焓的估算方法已有文献报道的有:定标粒子理论[3]、GIPF法(General Interaction Properties Function method)[4]、分子拓扑法[5-11]、基团贡献法[12-16]和其他估算法[17-20]。其中,前4种方法都需要应用临界参数,根据物质的临界性质所提供的信息来估算物质的蒸发焓。这些方法虽然估算精度较高,但许多物质尤其是大分子物质在临界区附近热稳定性较差,临界性质的数据很难获得,这就使方法的适用范围受到很大的限制。且这些估算方法,没有热力学基本原理支持,分子结构、分子间相互作用力与宏观性质的联系也未做考虑,或仅从化学的角度考虑,或仅从工程的角度考虑。
本工作从热力学基本原理出发,利用Abraham线性溶剂化能量关系(LSER)模型得到一套分子描述符,并考虑到蒸发焓与温度的关系建立了估算物质蒸发焓的新函数方程。该方程形式简单,估算精度高,适用范围广,成功地将物质的宏观物理性质与分子微观结构结合起来,为有机物蒸发焓的计算提供了一种可供选择的方法。
1 实验部分
1.1 Abraham线性溶剂化理论模型
目前,定量结构与活性相关(QSARs)模型[21]已广泛应用于预测化学品的性质、筛选风险化学品以及提供优先监控对象。线性溶剂化能相关方法也被公认是一种有效的QSARs模式。Kamlet等[22]指出,化学品的许多性质都取决于溶质与溶剂间的相互作用,因而可通过LSER方程式预测化学品的物理化学性质。LSER方程式可表示为:系统性质=常数+造腔项+偶极项+氢键项,该方程式被称为线性溶剂化关系模型。利用Abraham分子结构参数E,S,A,B和Vx,建立一个新的线性溶剂化能模型[23]:
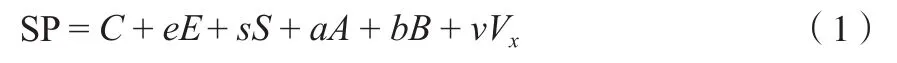
式中,SP 指系统的某项性质;E为过量摩尔折射率,用于衡量分子间色散作用;S为极化率,为一些极化作用的量度;A为氢键酸度,确切的应表示为ΣA,为总效应;B为氢键碱度,确切的应表示为ΣB,为总效应;Vx为McGowan特征分子体积[24],反映分子大小。
这套分子描述符数值唯一,意义明确,较容易获得,且不受物质范围限制,加上它与溶质的性质简单线性相关,无论准确度或精确度都很高,在性质估算方面具有很好的适应性[25-27]。
1.2 蒸发焓方程的提出
根据热力学基本原理,蒸发焓(ΔvapH)由两部分构成:

RTΔZV是蒸发过程中对气相所做的功。从微观角度看,蒸发过程是液体分子从溶液内部挣脱出来变成气体分子,而逸出分子不但需要克服与周围分子间的色散作用、取向作用、诱导作用等产生的范德华力,还需要挣脱分子间的氢键作用力、电荷转移作用力等。能量是各种分子间相互作用的总的反映形式,而分子结构、分子间的相互作用决定了各种物质的性质。物质的性质可以用分子描述符来表达。因此,蒸发焓中的蒸发内能可表示为:
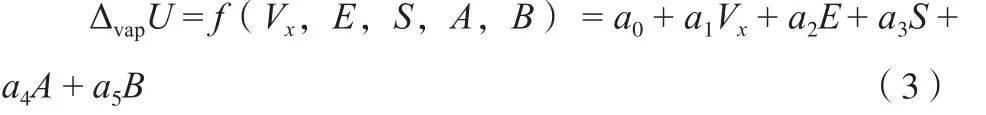
而 式(2) 中 的RTΔZv, 当 温 度 低 时,ΔZv≈1,此时ΔvapH与温度成线性关系;当温度高时,ΔZv=f(T)。文献结果表明,在较高的温度范围内,ΔvapH与温度的关系不再是线性,而是ΔvapH=hT n[28],这时ΔZv=f(T)应有式(4)关系:
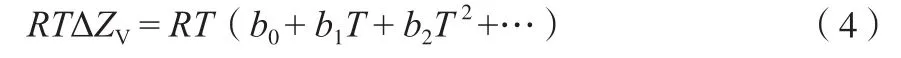
将式(3)和(4)代入式(2),导出估算不同温度下物质蒸发焓的新方程(5):

在有机物的熔点到沸点温度范围内,ΔvapH与温度的关系可表示为:

2 结果与讨论
2.1 数据的选取
本方法分子描述符较易由实验获得,也可由ADMEBoxes软件进行估算。本工作所取分子描述符主要来源于文献[23],有部分取自Sedov等[29-31]的文献值。本工作ΔvapH实验值数据全部来源于手册或发表的文献[1,32-44],其中,文献[1]的蒸发焓数据全部是由量热法获得。
2.2 方程相关性分析
对文献所选260种液体有机物,包括烷烃、烯烃、炔烃、卤代烃、醇、醚、醛、酮、酯和有机酸,共1 105个数据点,利用SPSS软件及EXCEL软件对实验数据及分子描述符进行回归,由最优化结果得到8个关联方程,方程(5)中各类物质对应系数结果见表1。从表1可看出,8个关联方程中各参数与ΔvapH的相关系数都达良好级以上,平均相对偏差值均低于5%。
为了更直观地表现出各关联方程适合数据体系的程度,对上述方程的ΔvapH数据分别做正态分布概率图,结果见图1。从图1可见,8类有机化合物在临界温度以下范围内的ΔvapH估算值的残差都接近正态分布,说明所拟合的方程可以用来估算对应有机化合物在不同温度下的ΔvapH。

表1 计算蒸发焓的新方程式(6)中的各项系数Table1 Coefficients in the new equation (6) for calculating evaporation enthalpy
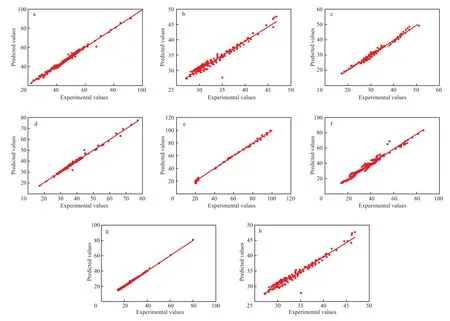
图1 8类有机化合物ΔvapH残差的正态分布Fig. 1 Normal distribution for ΔvapH residuals of 8 kinds of organic compounds.a Alcohols;b Halogenated hydrocarbons;c Ethers;d Aldehydes and ketones;e Acids;f Alkanes;g Alkenes and alkynes;h Esters
2.3 方程的外推适用性
为了验证各关联方程的外推适用性,用所得方程计算79种物质在298.15 K下的蒸发焓,并与各种数据手册常用的Watson方程计算结果进行比较,结果见表2。
从表2可以看出,在对链烷烃的估算中,Waston方法的预算精度要高于本工作所建方程,但从酮、醇、尤其是卤代烃的预算结果看,本工作建立的方程无论是估算温度范围还是估算精度都要优于Waston方法。
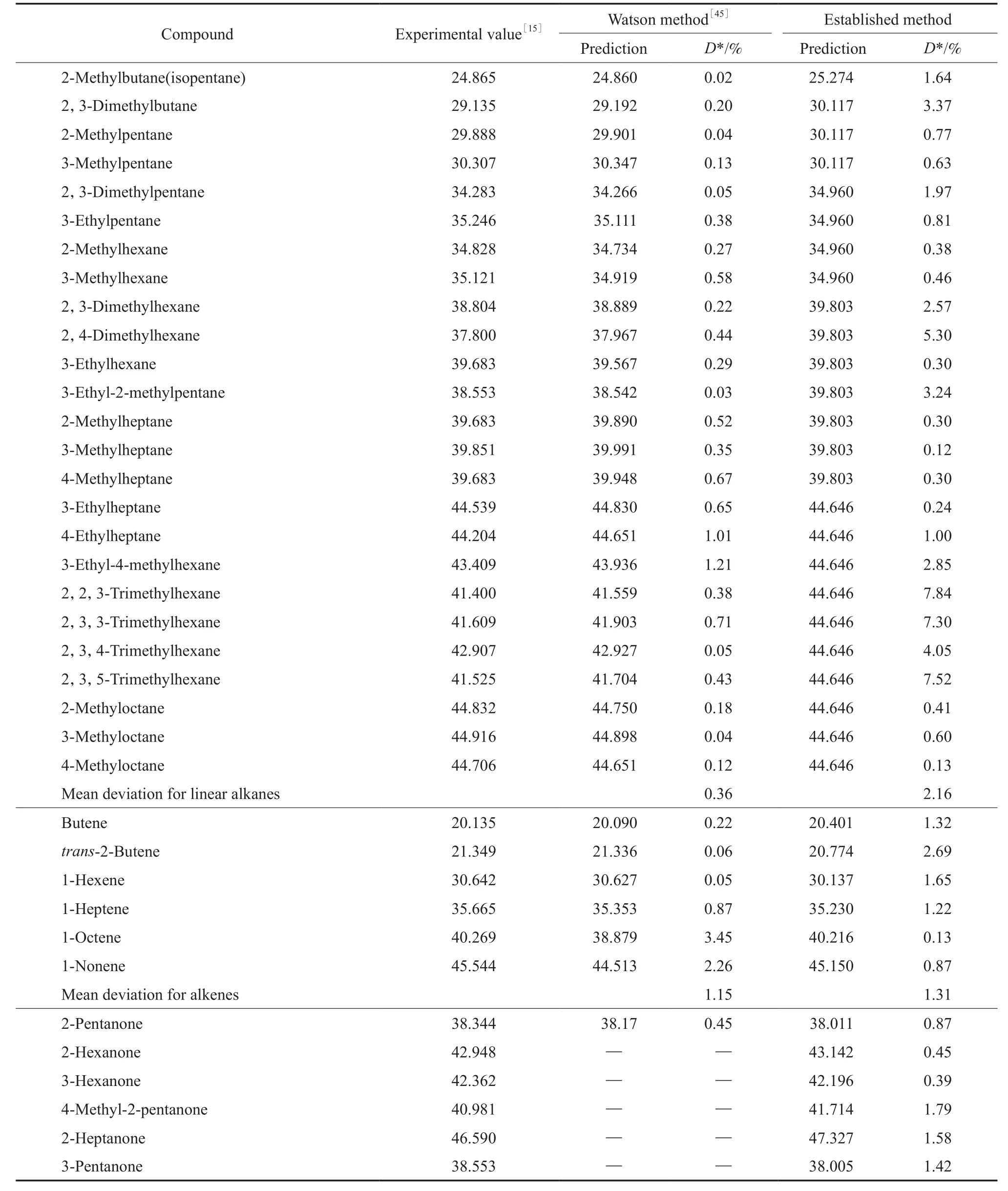
表2 79种有机化合物在298.15 K下ΔvapH的估算结果Table 2 The results of estimated ΔvapH for 79 organic compounds at 298.15 K
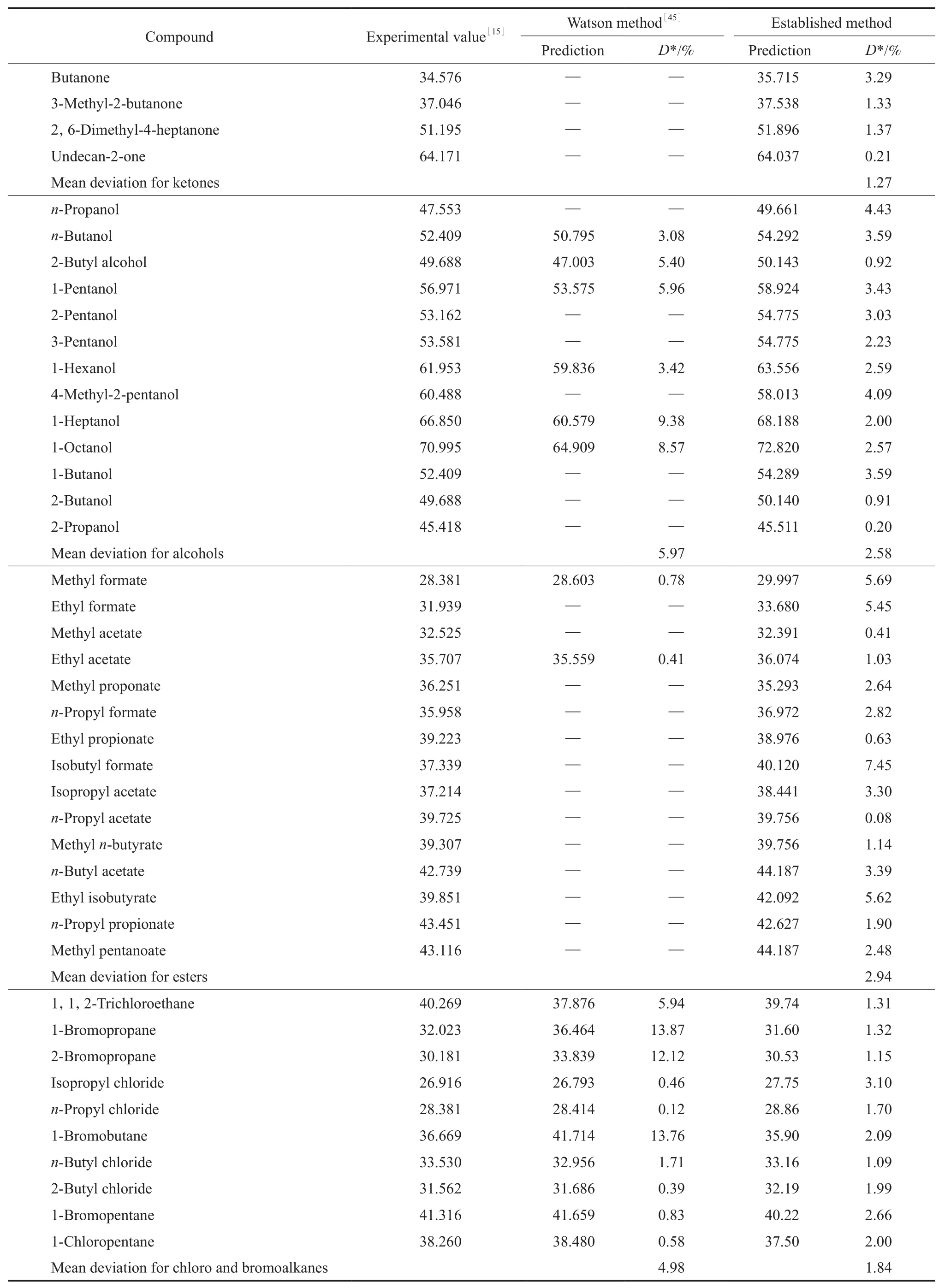
续表2
为了检验所建方程在其他温度下对蒸发焓的预测效果,用所建方程对部分物质在其他温度下的蒸发焓进行估算,结果见表3。从表3可看出,实验值和估测值基本相符,表明所建方程在其他温度下对蒸发焓的预测效果也较好,具有良好的普适性,成功地将物质的宏观物理性质与分子微观结构结合起来,为纯有机物蒸发焓的计算提供了一种可供选择的方法。
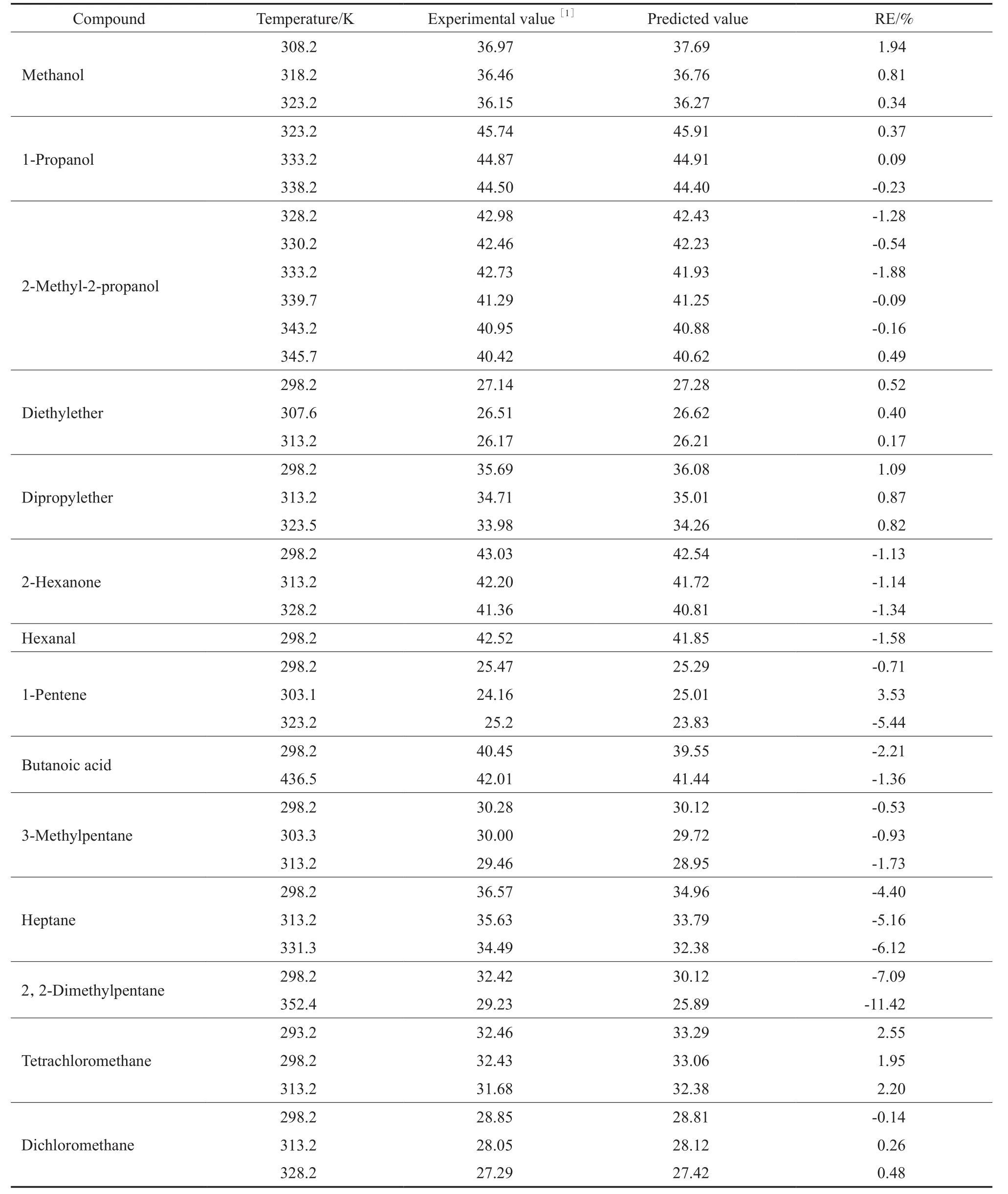
表3 有机化合物在不同温度下ΔvapH的估算结果Table 3 The results of estimated ΔvapH for organic compounds at different temperatures
3 结论
1)根据线性自由溶剂化能关系理论和经验方程,得到了蒸发焓的关联方程,方程的可预测温度范围广,对临界温度以下的蒸发焓都有良好的预测精度。从方程外推适用性分析,具有良好的普适性。
2)所得蒸发焓的关联方程对烷烃预测结果不理想,而对酮、醇,尤其是卤代烃的预测无论是估算温度范围还是估算精度都要优于Waston方法。
符 号 说 明

