蛋白酶体抑制剂Marizomib及其合成策略
2020-03-31吉胡瓦马宇衡
吉胡瓦,盛 华,马宇衡,布 仁
(內蒙古医科大学,內蒙古 呼和浩特市 010000)
1 泛素—蛋白酶体系统与26S蛋白酶体
泛素(ubiquitin)是一种存在于所有真核生物(大部分真核细胞)中的小蛋白。泛素由76个氨基酸组成,分子量大约8.451 kDa。它的主要功能是标记待水解蛋白质,使其被26S蛋白酶体降解[1]。泛素化(Ubiquitination/Ubiquitylation)是指泛素分子在一系列特殊的酶作用下,将细胞内的蛋白质分类,从中选出靶蛋白分子,并对靶蛋白进行特异性修饰的过程。这些特殊的酶包括泛素激活酶(Ubiquitin-activating enzyme,E1),结合酶(Ubiquitin-conjugating enzyme,E2)、连结酶(Ubiquitin-protein ligase,E3)和降解酶(Deubiquitinating enzyme,DUB)等。它们共同构成了泛素-蛋白酶体系(Ubiquitin-proteasome system,UPS)[2-5](图1)。泛素-蛋白酶体系统介导了真核生物体内80%~85%的蛋白质降解,该蛋白质降解途径具有依赖 ATP、高效、高度选择性的特点。

图1 泛素-蛋白酶体系统
UPS的核心是26S蛋白酶体,它可特异性识别泛素标记的蛋白质将它们水解成短肽。26S蛋白酶体形状如同一个圆桶,由两个19S蛋白酶体基底和一个20S蛋白酶体核心构成。由泛素标记的长肽经19S蛋白酶体识别后进入20S蛋白酶体核心被分解为短肽后再经另一端19S蛋白酶体释放[6-7]。20S蛋白酶体是由两个外层α环和两个内层β环堆叠而成的中空圆桶状亚复合体,每一层环由7个密切相关亚基组成,可表示为 α1-7,β1-7,β1-7,α1-7。蛋白酶体活性位点位于20S蛋白酶体内腔,由β亚基Thr1形成独特的单个残基催化位点,7种β亚基中有3种由于Thr1的存在具有催化活性:β1表现胱冬肽酶样(caspase-like, C-L)活性,β2表现胰蛋白酶样(trypsin-like, T-L)活性,β5表现胰凝乳蛋白酶样(chymotrypsin-like, ChT-L)活性并主要负责底物降解。26S蛋白酶体降解异常或错误折叠的蛋白质为细胞提供了控制蛋白质质量的机制,该机制与癌细胞紧密相关,癌细胞比正常细胞更高的速率增殖,因此表现出更高的蛋白质合成和降解速率,阻断26S蛋白酶体功能会使不需要的蛋白质积累,从而导致癌细胞死亡[8-9]。
2 Marizomib作用机制与构效关系
与第一代蛋白酶体抑制剂硼替佐米(Bortezomib)不同,Marizomib属于非肽类蛋白酶体抑制剂,对20S蛋白酶体的所有三个催化位点都具有很强的抑制活性(IC50:CT-L(3.5±0.3);T-L(28±2);C-L(430±34)nM)[10]。由于其结构分别在C-2,C-3和C-4处被氯乙基,甲基,环己-2-烯基甲醇基取代,所以将Marizomib分类为蛋白酶体抑制剂的β-内酯-γ-内酰胺类药物,这是一类来源于微生物及其衍生物的天然产物,包括Omuralide,Marizomib和Cinnabaramides。这些低分子药物对20S蛋白酶体有显著的特异性[11],甚至可与肽类抑制剂相媲美。Marizomib相比于其他β-内酯-γ-内酰胺双环结构化合物表现出更强的效价,使其成为了第二代蛋白酶体抑制剂研究热点[12-13]。通过Marizomib与20S催化颗粒的共晶体结构验证[14],Marizomib作用机制为不可逆的亲核攻击20S蛋白酶体(图2)。首先,内酯环的羰基与20S蛋白酶体活性位点N末端Thr1Oγ残基形成酯键,然后由Thr1上的NH2催化C-3O亲核取代氯化物,形成不可逆转的稳定的四氢呋喃环,最终对蛋白酶体有效且持久的抑制。
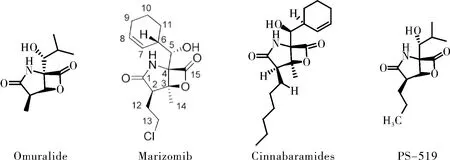
图2 β-内酯-γ-内酰胺类药物

图3 Marizomib作用机制
构效关系研究表明,内酯环是Marizomib的关键药效基团,在维持对蛋白酶体抑制活性方面起着至关重要的作用。同时氯乙基也是必要基团[14-16],当氯乙基被未卤代的基团取代会导致Marizomib对20S蛋白酶体的抑制活性降低,而氯离子被其他卤族元素取代时,仍有良好的活性(如碘乙基,IC50=(2.8±0.5)nM;溴乙基,IC50=(2.6±0.4)nM;氟乙基,IC50=(9.2±10.2)nM),这主要是由于其他卤素的离去能力也能形成高度稳定的四氢呋喃环[15]。同时在该位置上的其他离去基团,例如O-Ms(甲基磺酰基,IC50=(4.3±0.8)nM)和O-Ts(甲苯磺酰基,IC50=(2.5±0.4)nM),也产生了等效的抑制活性[16]。同时,C2氯乙基位置的立体化学结构对于活性也很关键[17],与Marizomib相比,氯乙基位置差向异构体活性衰减超过100倍[IC50=(330±20)nM]。环己烯环有利于Marizomib与蛋白酶体结合口袋之间的相互作用,在此位置基团的改变可能会导致效力降低和对蛋白酶体三个蛋白酶解位点的选择性变化。同时大分子烷基如环戊基,环戊-2-烯基和(2S,3R)-环氧环己基取代环己烯基比小分子烷基和芳族取代基取代环己烯基活性更好一些[17]。此外,C3位置甲基和C5位置的羟基是两个不可或缺的抗肿瘤活性基团,当被C3-Et(IC50=(2100±100)nM)或C5-O(酮,IC50=(8200±600)nM)取代导致>1000倍活性降低[17]。活性的降低与仅容纳C3-Me大小蛋白酶体活性位点口袋以及C5-OH与蛋白酶体之间形成氢键密切相关。
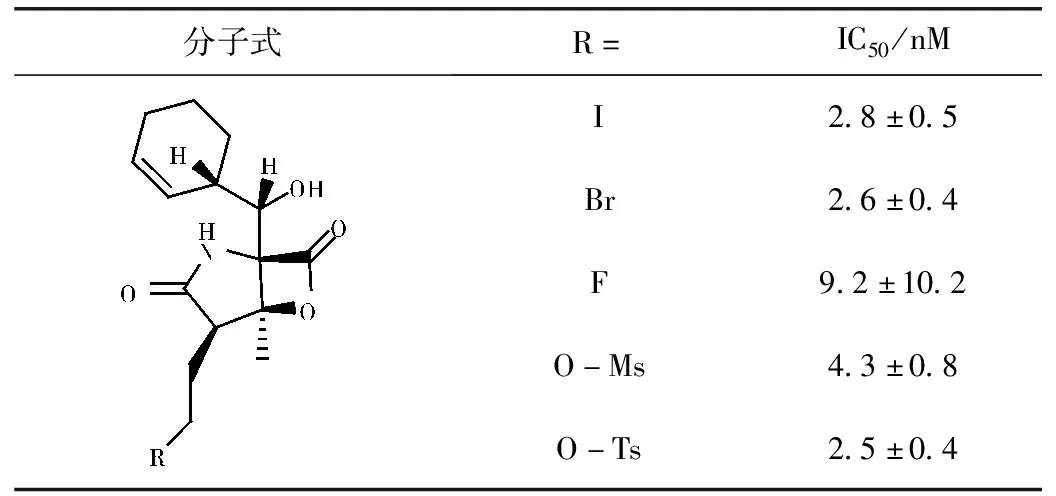
表1 当氯离子被其他离去基团取代时的活性[17]
3 Marizomib生物发酵法
Marizomib生物发酵法是目前生产Marizomib的主要方式,加州大学圣地亚哥分校Fenical研究小组研究出最初的发酵条件和生产菌株(S.tropicaCNB476),但在培养瓶中每升培养液仅能生产几毫克Marizomib,无法支撑临床试验所需的药物用量[18]。而后Nereus Pharmaceuticals研究了新的发酵条件,改善了培养基成分从而将产量由4 mg/L提升到了450 mg/L。
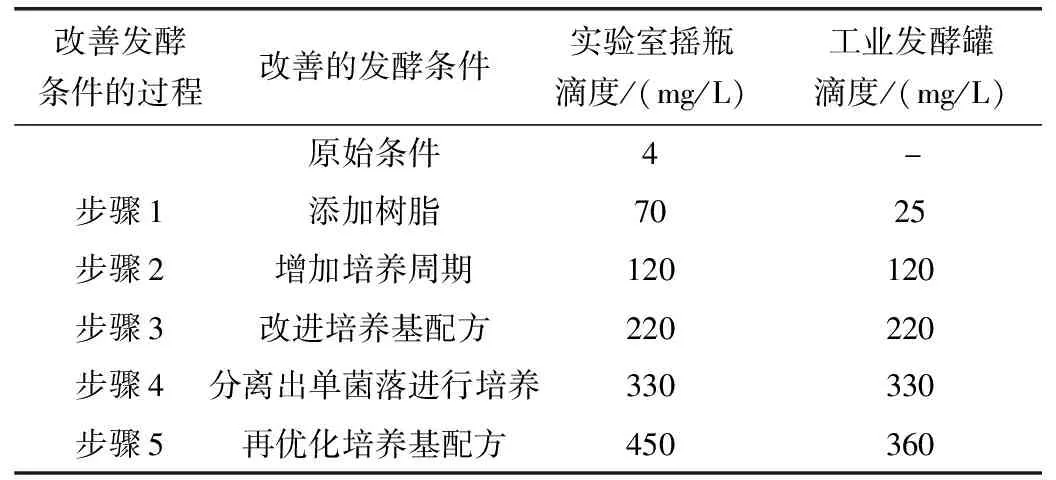
表2 通过改善发酵条件,使得Marizomib产量逐步上升[21]
因为研究表明,在发酵中添加树脂会使代谢产物的产量增加[19],所以最初提高Marizomib产量的关键是在培养液中添加固体树脂(表2步骤1)。通过在发酵液中加入固体树脂以结合和捕获Marizomib,用来克服水溶液中β-内酯环的不稳定性[20],在初步研究中,树脂的加入使产量提高了18倍(表2)。由于野生型菌株通常包含会产生Marizomib类似物的细胞群,所以使Marizomib产量提高的第二个关键是NPS21184菌株的分离,这是一种来自菌株CNB476无突变和遗传操作的单菌落分离株(表2步骤4)。除了能得到更高的产量,菌株NPS21184产生的脱氯类似物比亲代菌株CNB476少三倍,这对于之后Marizomib的纯化有重要意义。再经过改善培养基组成和解决发酵罐腐蚀问题后,Marizomib滴度在实验室摇瓶中达到了450 mg/L,在工业发酵中达到了360 mg/L,为Marizomib进入临床试验提供了药物基础。
4 不对称合成Marizomib
4.1 Corey不对称合成(-)-Marizomib
Corey及其同事在2004年第一个报道了对映选择性合成Marizomib[22],反应以(S)-苏氨酸甲酯为原料,与4-甲氧基苯甲酰氯进行N-酰化后,通过p-TsOH催化的环化反应得到相应的恶唑啉2。用ClCH2OBn进行立体选择性烷基化有效引入C-5,同时可以使C-4具有所需的手性,得到化合物3。用NaBH3CN-HOAc使恶唑啉开环,得到PMB保护的4。在TMS保护后,用丙烯酰氯进行选择性N-酰化并进行酸化处理,从而得到了具有连续的3碳C-1/C-2/C-12结构的化合物,随后Dess-Martin反应得到5。继续由奎宁环碱催化,使5分子内进行BaylisHillman反应,得到目标化合物内酰胺7与异构体6(9:1)。BrCH2Si(CH3)2Cl与三丁基氢化锡介导的自由基反应后与7顺式融合环化为β-内酰胺8。裂解苄基醚,将所得的醇氧化,得到关键的中间体醛9。使用Knochel方法将2-环己烯基与甲酰基碳连接得到化合物10并形成所需的C5和C6中心,这种方法在非对映选择性烯丙基化的同时具有高立体选择性(20:1)。用TamaoFleming氧化和脱保护使10脱去甲硅烷,得到11,在接下来三步操作中,将11转化为目标化合物(-)-Marizomib产率65%。Corey等首次通过全合成法获得了(-)-Marizomib,为之后Mairizomib的全合成指明了道路,具有里程碑式的意义。

图4 Corey不对称合成(-)-Marizomib
4.2 Marx不对称合成
Marx在2018年报道了以氧化自由基环化作为关键步骤合成(-)-Marizomib[23]。首先将左旋樟脑磺内酰胺12作为保护基与NaH反应后连接丁烯酰氯得到化合物13,将其连接溴乙酸叔丁酯得到白色固体14,产率为88%。再用过氧化氢溶液脱去12得到化合物15。下一步用T3P(propanephosphonicacidanhydride)连接16得到环化底物17。下一步是整个路线的关键步骤,将17使用Mn(OAc)3和Cu(BF4)2在乙腈中加热至80 ℃进行环化得到[3.3.0]-双环γ-内酯(±)-18,收率为50%~60%。后来他们发现溶剂乙腈中加入微量的水,比例为5:3时,双环内酯(±)-18收率最佳,能达到71%。分离纯化出(+)-18,将二硒二苯用NaH还原,再与(+)-18反应得到相应的羧酸19。将19转化为相应的酰氯,然后用三叔丁氧基氢化铝锂还原,得到相应伯醇20。使用高碘酸钠将20氧化为21,继续与苯硒基溴化物和四氟硼酸银反应,使21内酯化并脱去叔丁酯以90%的产率得到γ-内酯22。羧酸转化为相应的PMB酯23,在自由基条件下将苯基硒基还原为24。DMP与24反应后加入环己烯基溴化锌得到26。DIBAl-H与26反应,再添加甲醇和过量的硼氢化钠后得到三醇27。使用BOPCl形成β内酯,并用二氯三苯基磷进行氯化,得到Marizomib。
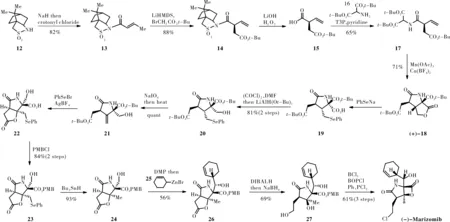
图5 Marx不对称合成(-)-Marizomib
4.3 Logan不对称合成

图6 Logan不对称合成(-)-Marizomib
Logan等在2014年发表了制备合成(-)-Marizomib关键中间体24,8步产率19%[24],并具有对映选择性。首先由γ-丁内酯25同二硒二苯,硼氢化钠反应生成化合物26。连接恶唑烷酮27保护,引入烯丙基得到29。脱去恶唑烷酮,进行Schotten-Baumann反应引入氨基丙二酸酯30生成酰胺31。酰胺31氧化消除硒化物得到对映体富集的环化底物(-)-32e。进行环化反应以65%的收率得到双环γ-内酯(-)-33e,将其进行臭氧分解并还原为醇34。继续用Reddy之前报导的方法继续进行即可得到(-)-Marizomib[25]。
4.4 Satoh不对称合成
Satoh[26]在2011年报导了由4-戊烯酸14步即可对映选择性合成(-)-Marizomib,总收率达到19%。该合成的特征在于利用手性助剂的立体选择性烷基化,形成吡咯烷和形成内酰胺时运用钌催化氧化。先将4-戊烯酸35与已知的手性恶唑烷硫酮的缩合,使用环状原酸酯进行烷基化得到单一构型的产物,还原除去手性助剂后,与丙二酸二甲酯和NaBH3CN反应,得到化合物37。将胺甲酰化然后脱去缩酮保护进行分子内环化,得到38a,38b,39的混合物,乙酸乙酯重结晶得到38a。用臭氧分解38a,然后进行酸化处理后得到双环产物40,除去甲酰基得得到了胺41a和41b的非对映异构体混合物。将混合物同时用2mol%钌催化剂氧化,得到极性较小的42a和极性较大的42b。纯化出42a并将其甲酯转换为苄酯后成为43,接下来43受位阻较小的苄酯被NaBH4选择性还原成为醇44。用TMS保护醇,再在内酰胺上引入Boc基团,脱去TMS后成为45。使用Dess-Martin将醇氧化成醛以引入环己烯基部分,得到46。在酸性条件下,脱去Boc基团同时内半缩醛向丙二醇的转化,然后用NaBH4还原甲酯得到三醇47。最后,使羧酸脱保护进行β-内酯化,然后对伯醇进行氯化得到(-)-Marizomib。

图7 Satoh不对称合成(-)-Marizomib
5 结 语
自从硼替佐米被FDA批准上市以来,己开发许多蛋白酶体抑制剂作为抗癌药。Marizomib作为第二代蛋白酶体抑制剂已进入Ⅲ期临床试验,其对蛋白酶体抑制有效且持久,使Marizomib有希望被批准上市成为某类癌症患者的希望。虽然工业上运用生物发酵法量产Marizomib,但化学合成法对结构改造及构效关系的研究仍有重要意义。
