T淋巴细胞激活蛋白家族2对白细胞的调控作用
2020-03-31庄学玲熊礼宽
梁 卉 王 尊 庄学玲 熊礼宽
T淋巴细胞激活蛋白家族2基因(the linker for activator of T-cells family member 2, LAT2) 由Doyle等于2000年在人类和小鼠编码Williams Beuren综合征关键位点5蛋白(Williams Beuren syndrome critical region 5, WBSCR5)区域内发现,命名为WBSCR15。2002年Brdicka等利用体外激酶分析,在髓细胞鞘糖脂富集膜(glycophingolipid-enriched membrane, GEMs)蛋白区域蛋白中发现并命名为非T淋巴细胞激活蛋白(non-T-cell activator linker, NTAL)。2003年Janssen等成功克隆了WBSCR15序列,由于其在B淋巴细胞中表达和发挥作用,因此命名为B淋巴细胞激活蛋白(linker activator for B-cells, LAB)。2006年人类基因组命名委员会根据蛋白结构特点,将其命名为LAT2。
LAT2是一个跨膜转接蛋白(transmembrane adaptor proteins, TRAPs),具有多个酪氨酸磷酸化位点,在后续通路中发挥支架和信号传递功能。LAT2主要表达于髓样细胞和淋巴样细胞中,如肥大细胞、B淋巴细胞、自然杀伤细胞(natural killer cells, NKs)、嗜碱性粒细胞和单核细胞等。LAT2磷酸化后可直接或间接捕获细胞质内信号分子,包括生长因子受体结合蛋白2(growth factor receptor-bound protein 2, GRB2)、非七激酶子同源物1(son of sevenless homolog 1, SOS1)、GRB2相关连接蛋白1(GRB2-associated binding protein 1, GAB1)、泛素连接蛋白(casitas B-lineage lymphoma, C-CBL)和Vav蛋白等,从而发挥其生物学效应。
一、LAT2基因克隆和表达
LAT2基因差异性表达:(1)与细胞状态有关,活化后的T细胞、NK细胞和衰老细胞中,LAT2表达上调[2,3]。(2)在细胞成熟过程中呈动态改变,正常B淋巴细胞的成熟过程中,LAT2的表达量增加;正常T细胞成熟过程中,LAT2的表达量降低[1]。(3)髓系细胞分化过程中呈多元态势,LAT2在髓系干细胞中低表达,向粒细胞、单核细胞和红细胞分化时,LAT2的表达量先增加(第7天表达量最高),随后降低[4]。但另一项研究发现,LAT2的表达趋势与分化方向有关,髓系细胞分化为单核细胞时,LAT2表达逐渐上调;而向粒细胞分化时,LAT2表达逐渐下降[1]。(4)不同类型白血病细胞的LAT2表达不同,单核系来源的急性髓细胞性白血病(acute myeloid leukemia,AML)细胞中,如AML-M4、AML-M4eo和AML-5,LAT2表达上调;而在粒系来源的AML细胞中,如M2和M3,LAT2表达下降;而当存在t(8,21)形成融合蛋白RUNX1/RUNX1T1时,LAT2不表达[1]。在急性淋巴细胞性白血病(acute lymphoblastic leukemia of childhood, ALL)原始细胞中也发现LAT2的表达量增加,儿童前体B-ALL细胞中LAT2 mRNA的表达量约为正常B淋巴细胞的4倍[1]。(5)LAT2表达量还与T-ALL对药物的敏感度和疾病预后有关,LAT2高表达患者对泼尼松更敏感,而LAT2低表达患者对泼尼松不敏感。(6)LAT2表达水平还可用于肿瘤的生存期预测和预后评估,LAT2表达增高与星形胶质瘤、克罗恩病、卵巢癌、肺癌和乳腺癌的生存期和预后呈正相关[3]。
目前LAT2表达的调控机制尚不清楚。研究发现,LAT2基因是AML/ETO的靶基因(与LAT2基因的内含子3区域结合)。当细胞中存在AML1/ETO时,LAT2表达明显下降;敲除AML1/ETO后,LAT2表达上调,提示AML1/ETO可直接调控LAT2的表达[5]。免疫共沉淀发现AML1/ETO与LAT2结合后,LAT2基因的活性组蛋白修饰降低,而非活性组蛋白修饰增加。特别是在LAT2转录结合区域上游的1.5kb处发现有非活化组蛋白的富集,以上均提示AML1/ETO与LAT2结合后,通过组蛋白表观修饰来抑制LAT2的表达[5]。
二、LAT2蛋白结构和功能
LAT2相对分子质量为(25~30)kDa,为第Ⅲ类完整跨膜蛋白,具有典型的跨膜蛋白结构,包含短的胞外区(1~5),含CxxC棕榈化位点的跨膜区(6~26)和长的胞质区(27~243,图1)。LAT2本身不具有催化活性,但可与细胞膜上的GEMs相关联,其作为一个支架蛋白,募集具有SH2结构域的转导分子加入受体信号复合物中(信号转导小体)。
目前的研究发现LAT2包含3类重要位点,即棕榈化位点、酪氨酸(tyrosine, Tyr)磷酸化位点和溶蛋白酶裂解位点(图1)。
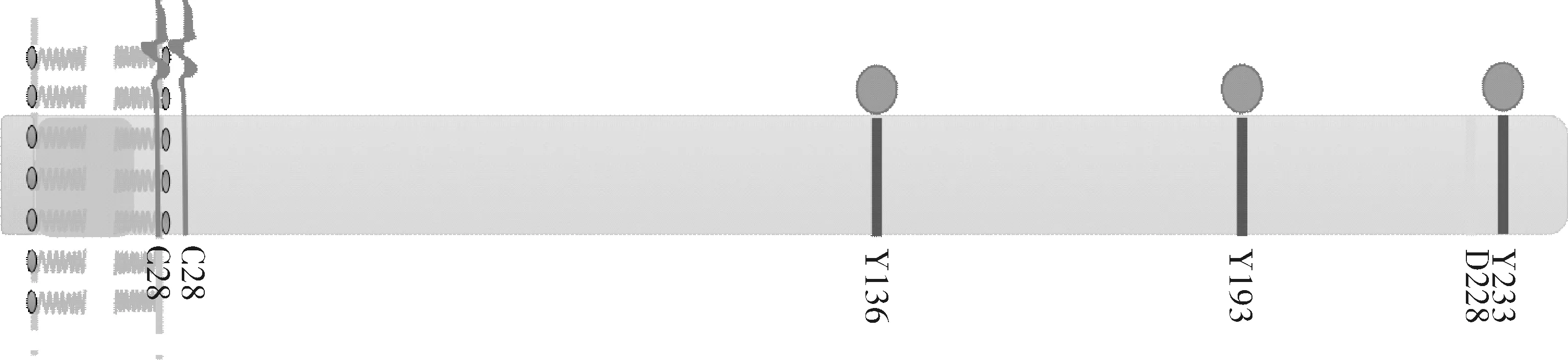
图1 LAT2蛋白结构示意图
LAT2蛋白包含243个氨基酸,具有胞外区(1~5)、跨膜区(6~26)和胞质区(27~243)。C25和C28是其棕榈化位点,是该蛋白在脂筏上定位的重要结构。包含10个预测的酪氨酸磷酸化位点,其中Y136、Y193和Y233被证实磷酸化与其生物功能有关。D228为溶蛋白酶裂解位点,其裂解后LAT2的磷酸化水平大大下降,生物学功能也降低。
LAT2蛋白的棕榈化位点位于第25和28个氨基酸(C25-V-R-C28),在GEMs上的定位和支架形成过程中具有重要作用,是其发挥生物学功能的必要条件,也可通过竞争性占位发挥作用[6]。当给予烷化溶血磷脂(alkylphospholipid, APL)药物时,由于APL可与细胞膜上的胆固醇结合,竞争性占位GEMs,使得LAT2的棕榈化位点无法锚定在GEMs上,最终被蛋白酶水解[6]。LAT2的竞争性占位还体现在树突状细胞(dendritic cells, DCs)的抗感染反应中[7]。
LAT2在膜受体介导的信号转导的启动阶段起着重要的作用,该过程依赖于胞质区的Y残基磷酸化。LAT2的胞质区中共包含10个Y残基位点(Y40、Y58、Y84、Y95、Y110、Y118、Y119、Y156、Y183和Y233),生物信息预测发现Y95、Y118、Y119、Y156、Y183和Y233可能与胞质中的转接蛋白Grb2的SH2区域结合,Y233个位点与LAT上的GADS SH2区域结合的位点同源。实验研究发现,LAT2末梢的3个酪氨酸残基(Y136、Y193和Y233)是其主要的功能域,当酪氨酸残基突变为苯丙氨酸(phenylalanine, Phe)时,LAT2磷酸化程度,与Grb2的结合作用和Ca2+流通均发生降低(单独突变时)或消失(任意2个位点突变或3个位点均突变时)[7]。
LAT2的磷酸化依赖于Src家族和c-kit。在293T细胞系中,Src激酶家族成员,如Lyn、Lck、ZAP-70和Sky均直接参与LAT2的磷酸。在B淋巴细胞中,Sky活化可磷酸化LAT2[5,8]。在肥大细胞中,FcεRI介导和SCF介导LAT2磷酸化分别由Src家族和c-kit调控,Lyn、Sky和Kit选择性磷酸化不同的LAT酪氨酸残基位点,但目前尚未有研究探讨其差异细节[8]。
LAT2溶蛋白性裂解导致细胞内信号转导的效率降低或终止,可能是细胞内信号转导负调控的机制之一。Arbulo-Echevarria等[9]发现在Fas或十字孢碱(staurosporine)作用下,LAT2从D228处裂解为约22kDa蛋白,CD3刺激后,LAT2整体磷酸化水平、与Grb2的结合能力以及PLC-γ的磷酸化均下降,LAT2溶蛋白性裂解可降低或终止细胞内信号转导。但截断的LAT2不影响Erk的活性水平。因此,溶蛋白性裂解可能是活化T淋巴细胞中Fas促进细胞死亡和终止TCR介导信号转导的机制。
这名字一听就是有来历的,藏着一个古老的传说。但我无意探寻来龙山名的传说。我想探寻的,是一条上山的路,想沿着山路爬到山顶上去。
LAT2磷酸化与溶蛋白性裂解呈负相关。LAT2溶蛋白性裂解后,丢失了重要的磷酸化位点Y233,LAT2整体磷酸化水平明显下降。过钒酸钠可促进LAT2的磷酸化,通过预先加入过钒酸钠可抑制Fas介导的LAT2裂解。此外,将D228附近的两个磷酸化位点(S223和Y233)突变后,加入过钒酸钠抑制裂解的作用消除,提示LAT2的磷酸化作用可抑制蛋白裂解作用。可能的机制是LAT2磷酸化导致蛋白结构发生改变,隔断蛋白酶和裂解位点,此外,磷酸化后引入带负电荷的磷酸盐也可抑制裂解[9]。
三、LAT2在细胞中的作用
1.T淋巴细胞:T淋巴细胞分化成熟过程中,LAT2表达量逐渐降低,初始T细胞中LAT2不表达,活化后,LAT2表达[1,5]。LAT2-/-小鼠的T细胞正常成熟分化,但极度活跃,TCR刺激后,T淋巴细胞中的ARK活性,Ca2+的分泌和细胞因子的产生均增强。由于T细胞的过度活化,LAT2-/-小鼠极易发展为自身免疫性疾病[5]。当LAT2过表达时,ERK和AKT的活性降低,抑制T细胞的活性。综上所述,LAT2在活化T淋巴细胞中的负调控作用可能是T细胞维持正常功能的机制之一。
LAT2在T淋巴细胞中的作用机制目前存在两个假说。一是竞争性抑制LAT。LAT与LAT2基因的序列相似,编码的蛋白质结构类似,被认为具有相同的进化起源,在细胞中呈现出相互补偿的表达模式。在LAT2-/-小鼠的T淋巴细胞中,发现LAT的磷酸化水平增高,而在超表达LAT2的T淋巴细胞中,LAT的磷酸化水平降低。LAT2缺陷的T淋巴细胞膜脂筏(lipid rafts)上定位了更多的LAT,以上均提示LAT2和LAT在T淋巴细胞中表现为拮抗作用[5]。该假说部分解释了LAT2的负调控机制,但无法解释生理状态下LAT2和LAT的相互作用。二是LAT2可能通过溶蛋白酶裂解作用抑制T淋巴细胞的活化。Arbulo-Echevarria等[9]研究发现,在Fas或十字孢碱作用下,LAT2从D228处裂解为约22kDa的蛋白,裂解后的LAT2蛋白N端仍锚定在脂筏上,但是其整体的磷酸化水平,捕获GRB2能力及PLC-γ的磷酸化水平均大幅度降低,进而抑制TCR介导的信号传递。但是截断的LAT2蛋白可维持足够的ERK的活性水平,促进了Fas介导的细胞死亡,进一步抑制了T淋巴细胞的活性和恢复T淋巴细胞的数量,防止T淋巴细胞过度活化。
2. B淋巴细胞:LAT2在B淋巴细胞整个生命周期中存在差异表达,未成熟的B淋巴细胞中表达较低,细胞成熟后LAT2表达上调,浆细胞中也表达。
LAT2不是B淋巴细胞发育过程中的关键蛋白,表现为LAT2单基因敲除(knock out, KO)小鼠中,仅发现边缘带B细胞数量减少,而其他类型B淋巴细胞发育并未受影响[3]。LAT2可能参与BCR介导的反应,包括Ca2+流通和BCR内吞作用。在LAT2 KO小鼠中,BCR刺激可导致Ca2+流通轻微增加,提示LAT2在B淋巴细胞中的负调控作用。但是在DT-40(成熟的鸡B淋巴细胞系)细胞中,超表达LAT2导致pro-BCR诱导的Ca2+流通增加,突变后的LAT2不影响Ca2+流通,提示LAT2对B淋巴细胞的正调控作用。LAT2对B淋巴细胞中Ca2+流通的差异作用机制还不清楚,可能与诱导物(BCR或pro-BCR)的差异、B淋巴细胞来源以及细胞成熟程度不同有关。
LAT2参与BCR的内吞作用,Malhotra等[10]发现,BCR刺激B淋巴细胞后,LAB发生磷酸化。磷酸化后的LAT2分别与GRB2-dynamin(驱动蛋白)和Vav蛋白结合,形成dynamin-GRB2-LAT2-Vav复合物,该复合物中的Vav活化小分子GTPsaes Rac1/2,进而发挥BCR内吞作用和向T细胞递呈抗原的作用。由于Vav和Rac是BCR内吞作用的关键蛋白,而LAT2缺陷的B淋巴细胞中BCR介导的Vav磷酸化和Rac活化减低,提示LAT2参与BCR的内吞作用。但是在LAT2 KO小鼠中,BCR的内吞作用有所减少,可能存在不包含LAT2的其他途径或其他支架蛋白可替代LAT2的部分作用。
综上所述,LAT2的缺陷会部分影响BCR的内吞作用,因此,LAT2缺陷的B淋巴细胞介导的体液免疫可能减少而非完全丧失。在LAT2 KO和野生型小鼠中,也未发观察到B淋巴细胞对TNP-OVA的抗体反应性存在显著性差异。
3.肥大细胞:不同来源和干预方式获得的肥大细胞在受到免疫球蛋白E受体(immunoglobulin E receptor, FcεRI)刺激后,LAT2的调控方向不同。骨髓来源肥大细胞(bone marrow derived mast cells, BMMCs)中,LAT和LAT2均有表达,LAT单基因KO小鼠和LAT、LAT2双基因KO小鼠的BMMCs对FcεRI刺激均表现为低反应性,但是,双基因KO小鼠BMMCs的反应性更低,提示LAT缺陷时,LAT2正向调控FcεRI信号。此外,在LAT2 KO小鼠的BMMCs和人肥大细胞LAT2单基因敲减(knock down, KD)模型以及嗜碱性粒细胞性白血病细胞中发现脱颗粒作用受损,提示在LAT存在下,LAT2也正调控肥大细胞功能[11]。但在其他研究小组发现LAT2 KD小鼠的 BMMCs对FcRI刺激表现为高反应性:ERK活性增强,Ca2+流通增加,脱颗粒和细胞因子产生增加,Polakovicova等[11]在LAT2 KO和KD的BMMCs中均观察到脱颗粒、Ca2+流通、趋药性增强,以及LAT和ERK磷酸化增强和丝状肌动蛋白解聚作用,提示LAT2负调控FcεRI信号。LAT2在肥大细胞中的相互矛盾的调节作用的机制还不清楚。有研究者认为,LAT2的负调控作用可能是通过竞争性抑制LAT来发挥的:在LAT2 KO的BMMCs中,存在抗原介导的PLC1、PI3K和ERK活性增强和Ca2+流通增强,同时,在这些细胞中,发现LAT的磷酸化增强。因此,这些高反应性表型体现的是LAT介导的反应的过度补偿导致的异常信号。此外,LAT2与LAT竞争性结合细胞膜上的脂筏也可能是导致其负调控作用的可能机制。而LAT2在活化的肥大细胞中的正调控作用可能与磷酸化后的LAT2结合了相应信号分子有关。
LAT2抑制前列腺素E2(prostaglandin E2, PEG2)介导的肥大细胞趋化性。与FcεRI不同,PEG2并不诱导LAT2的磷酸化,但LAT2缺陷的肥大细胞对PEG2的趋化性增强。PEG2刺激LAT2-/-肥大细胞后,AKT磷酸化增强,并促进了磷脂酰激醇3,4,5-三磷酸的生成。LAT2-/-静止型肥大细胞存在肿瘤侵袭性增加的特征,如ERM家族蛋白中抑制性苏氨酸磷酸化和β1整合素活性增加。加入全长的LAT2后,可恢复LAT2-/-肥大细胞对PEG2的趋化功能[12]。
LAT2具有维持肥大细胞骨架的功能。与野生型BMMCs比较,在抗原刺激下,LAT2-/-BMMCs的纤维蛋白附着减少、丝状肌动蛋白解聚作用增加和迁移性增加。该作用可能与GTPase Rho相关,LAT2对GTPase Rho活性具有正调控作用,当LAT2缺陷时,RhoA活性快速降低。而将LAT2转导至LAT2缺陷的细胞中,纤维连接蛋白附着和肌动蛋白的反应性均增加。因此,LAT2通过Rho和Rho相关蛋白激酶(Rho-associated protein kinase, ROCK)在肥大细胞骨架的维持中发挥重要的作用,表现为正调控肌动蛋白的聚合作用和细胞附着,以及负调控趋药性[13]。
4.粒细胞:LAT2在骨髓造血细胞中低表达,在造血细胞分化过程中,LAT2的表达发生改变[4]。Duque-Afonso等[1]在HL-60和NB4细胞系中发现,当使用全反式维甲酸诱导分化粒系细胞时,LAT2的表达下调,而当佛波酯诱导细胞系向单核细胞分化时,LAT2表达上调。健康人的CD34+骨髓细胞分离后,经体外细胞因子刺激诱导分化,LAT2表达先增高,在第7天(粒系/单核系)或第9天(红系)最高,随后降低[4]。此外,在髓细胞性白血病中,也存在LAT2表达异常。Duque-Afonso等[1]从不同FLATAB分型的急性白血病患者中分离的骨髓细胞中发现,LAT2在大多数原始白血病细胞中高表达,特别是在粒-单核细胞特性和淋巴细胞性白血病。以上均提示,LAT2可能参与髓细胞的成熟分化。
5.NK细胞:在LAT-/-和LAT-/-/LAT2-/-的小鼠中,NK细胞的数量增加和Ly49库的抑制性受体表达改变,提示LAT和LAT2参与NK细胞的发育[14]。由于Ly49库的改变与PLCγ的水平有关,而LAT和LAT2直接或间接与PLCγ相互作用,因此,LAT和LAT2可能通过PLCγ来参与Ly49库的调控。LAT-/-NK细胞保留溶解靶细胞的能力,同时靶细胞中Fc受体介导的ADCC功能正常。而在IL-2活化的LAT-/-/LAT2-/-的NK细胞中,DAP12介导的细胞毒性和IFN-γ的产生均丧失,表明LAT2可能补偿了LAT的功能。Whittaker等[14]发现NK细胞中至少存在ITAM-Sky-LAT2/LAT和ITAMZ-Zap70-LAT两种信号框,LAT正调控NK1.1功能,而LAT2对NK细胞起到负调控作用。NK细胞在IL-12刺激下,LAT表达下降,LAT2表达上调,结合基因敲除的功能研究结果推测活化的NK细胞作用主要与LAT2相关。
6.树突状细胞(dendritic cells, DCs):LAT2在DCs中表达,表达量略低于肥大细胞和巨噬细胞,参与DCs抗细菌和真菌感染的固有免疫应答。LAT2-/-缺陷小鼠中DCs的分化不受影响,但存在缺陷,表现为MHC Ⅱ类分子表达量下降。在LPS刺激下,LAT2-/-DCs中的ERK、JNK和p38活化增强,且LAT2-/-小鼠对LPS诱导的感染性休克呈高反应性,提示LAT2在DCs中负调控TLR4介导的信号的MAPK活性[7]。在其他细胞中,当细胞表面受体聚集后,LAT2磷酸化后参与细胞内因子的级联反应来发挥作用,但是当LPS刺激后,并未观察到DCs细胞中的LAT2磷酸化水平增加,同时,LAT2的5个酪氨酸磷酸化位点突变后,与野生型细胞中LPS介导的反应相似。上述实验表明,在DCs中,LAT2可能通过其他途径参与调控,可能与LAT2和其他分子在脂筏上的竞争性占位有关,需进一步研究证实[7]。在真菌感染过程中,磷酸化的LAT2通过抑制β-catenin向细胞核移动,进而促进IL12家族细胞因子的表达和IFN-γ的产生,来发挥抗真菌感染的免疫反应。LAT2-/-小鼠易被白色念珠菌感染,以及LAT2-/-DCs中的IL-12家族细胞因子表达下调,提示LAT2在清除真菌感染中发挥正调控作用[15]。
7.巨噬细胞:在单核细胞分化为巨噬细胞过程中,LAT表达下调而LAT2表达升高。与DCs相似,巨噬细胞中LAT2表达,而LAT不表达。研究发现,LAT2-/-巨噬细胞中,TREM-2介导的Erk1/2活性增强,Syk和c-CbL磷酸化水平增加,提示LAT2负调控TREM-2介导的Erk1/2活化,导致抗炎性反应增强。其可能的机制是LAT2被Syk磷酸化后,与Grb2结合,进而使Erk1/2活化,并招募c-Cbl。随后c-Cbl下调邻近的TREM-2信号,包括Syk的活化。
8.上皮细胞:LAT2在上皮细胞的炎性反应中正向调控ERK通路。当幽门螺旋杆菌(helicobacterpylori, HP)感染人结直肠癌细胞系HCA-7后,LAT2和淋巴细胞特异性络氨酸蛋白激酶相互作用膜蛋白的磷酸化水平增加。活化的LAT2通过Grb2的相互作用与c-Met-Grb2-Sos1蛋白复合物相互作用,Sos1活化Ras-Raf-1-MEK1/2-ERK通路,活化cPLA2。活化的cPLA2生成氨基酸,参与炎性反应的启动和传播。
四、展 望
LAT2是一个跨膜转接蛋白,主要存在于白细胞中。LAT2通过棕榈化位点、磷酸化位点和溶蛋白裂解位点发挥作用。LAT2蛋白通过棕榈化位点锚定在细胞膜表面,发挥占位和维持LAT2蛋白稳定的作用。磷酸化位点是LAT2的主要功能位点,当细胞受到刺激后,Src家族和c-kit磷酸化介导LAT2磷酸化,并将信号传递给下游蛋白,发挥负调控作用和(或)正调控作用。LAT2的溶蛋白裂解导致细胞内信号转导降低或终止,参与LAT2负调控作用。LAT2的功能研究主要集中于肥大细胞和淋巴细胞中,参与免疫反应。由于LAT和LAT2在结构上同源,在多种细胞中表达互补,且动态改变,因此,对于LAT2的功能研究,还要排除LAT的补偿作用。目前对于LAT2的功能研究多使用LAT2-/-模型,无法体现生理状态下LAT2的作用以及无法解释LAT2在细胞膜上占位引起的生物学改变。
严重的先天性中性粒细胞减少症(severe congenital neutropenia, SCN)是一种先天性骨髓衰竭疾病,遗传因素为其主要致病因素。一些致病基因(如ELANE、GFI1、CSF3R、WAS和HAX1等)突变导致髓细胞成熟分化障碍,使得髓细胞处于幼稚阶段。SCN具有急性髓细胞白血病的风险,被认为属于白血病前期。已有报道发现LAT2参与白细胞的成熟分化,且在髓性白血病中发现LAT2的表达异常,同时笔者在一个SCN家系中发现LAT2突变且与表型共分离,提示LAT2在SCN中的潜在作用。从 LAT2 的功能特性和患者人群遗传分析结果来看,LAT2基因可能参与了 SCN 的发病,但其是否为SCN新的致病基因及其在SCN中的作用和机制还不清楚,需要开展深入的研究证实。
