Ni2P的制备及含磷杂质对其催化DBT加氢脱硫性能的影响
2020-03-27林梦男孙志超王安杰
林梦男, 孙志超, 王 瑶, 王 伟, 石 川, 王安杰
(1.大连理工大学 精细化工国家重点实验室,辽宁 大连 116024;2.辽宁省高校石油化工技术与装备重点实验室,辽宁 大连 116024;3.银川能源学院,宁夏 银川 750105)
随着全球日益增长的能源需求以及随之而来的环境污染问题,高效清洁利用传统能源并寻找可持续、环境友好的新型能源成为解决这一问题的核心。其中,解决这一问题的关键是开发廉价、高效的催化剂。近年来,过渡金属磷化物(TMPs)催化剂已在冶金、加氢脱硫(HDS)、加氢脱氮、电化学、光催化、锂离子电池等多个方向表现出优异的性能。过渡金属磷化物种类繁多,其中Ni2P以其优异的类贵金属Pt性质受到了广泛的关注。Ni2P可通过多种镍源(如无机镍盐或有机镍源)和磷源(如单质磷、磷酸盐、亚磷酸盐或有机磷等)制备,且不同的前体会导致制备过程的差别[1]。例如,早期主要是通过剧毒的白磷或磷化氢作为磷源直接磷化合成;之后,Li等[2]以磷酸盐作为磷源,采用程序升温还原法(TPR)成功制备出Ni2P催化剂。该方法也成为制备金属磷化物最主要的方法[3-4]。然而,由于磷酸盐中P—O键键能较高(410 kJ/mol),Guan等[5]和Tian等[6]分别以次磷酸盐和金属磷硫化物代替磷酸盐,通过还原相对较弱的P—O键和P—S键来降低制备所需温度。但由于制备过程会导致磷挥发,因此样品需要在高于所需的金属/磷摩尔比条件下制备。这可能会导致部分杂质或磷物种附着在催化剂上,需要通过洗涤的方法除去。Guan等[7]以磷酸盐为磷源,在H2等离子体(PR)条件下按化学计量比(镍/磷摩尔比为2)制备出Ni2P催化剂。此外,还有以有机磷(TOP和TOPO等)作为磷源的合成法[8]和以红磷作为磷源的水热合成[9]方法等。不同方法制备的Ni2P催化剂,其反应活性表现出明显差别,如水热合成法制备的催化剂相比其他方法制备的催化剂表现出较差的加氢脱硫催化性能[10]。实验室前期研究中发现,在H2等离子体条件下,若以次磷酸盐代替磷酸盐,在镍/磷摩尔比为2的条件下无法成功制备出Ni2P催化剂,而是出现NH4NiCl3和Ni(PO3)2等杂质[11]。同时,制备过程中会出现NiCl2过度还原生成镍的情况,需要将等离子体气氛更换为N2,或增加NaCl、KCl等盐类抑制其过度还原[11]。也就是说,制备Ni2P过程中所使用的磷源可能并未全部生成Ni2P晶相,而是一部分以含磷杂质的形式存在,进而影响催化剂的性能。因此,深入探讨Ni2P制备过程中可能生成的含磷杂质,进而优化制备条件,对合成Ni2P催化剂至关重要。为避免过多因素的干扰,笔者选取氢氧化镍为镍源,次磷酸为磷源,系统考察了还原终压的升高速率、还原气速及还原终电压、还原时间对催化剂合成的影响,同时结合模型含硫化合物二苯并噻吩(DBT)的HDS反应结果及X射线衍射(XRD)、电感耦合等离子体发射光谱(ICP-AES)和傅里叶红外(FT-IR)的表征,进一步探讨了反应过程中含磷杂质的变化及其对催化剂催化性能的影响。
1 实验部分
1.1 原料
氢氧化镍(Ni(OH)2)、次磷酸(H3PO2,50%(质量分数)水溶液)、硝酸镍(Ni(NO3)2·6H2O)、磷酸氢二铵((NH4)2HPO4)、十氢萘,均为分析纯,中国国药集团产品;联苯(BP)、硫(S),分析纯,上海阿拉丁生化科技股份有限公司产品;石英砂(粒径380~830 μm),天津石英钟厂霸州化工分厂产品;H2(体积分数99.99%)、H2S/Ar(体积分数10%),中国科学院大连化学物理研究所提供。二苯并噻吩(C12H8S)由联苯和硫合成[12]。
1.2 H2等离子体(PR)法制备Ni2P催化剂
采用共沉淀方法制备体相Ni2P催化剂前驱体。将Ni/P摩尔比为1的Ni(OH)2和H3PO2分别溶于水中,并将H3PO2水溶液滴加到Ni(OH)2中,充分搅拌后在80 ℃下干燥12 h,得到以次磷酸为磷源的催化剂前驱体。将催化剂前驱体研磨,压片,破碎,过筛,得到粒径380~830 μm的颗粒备用。
取0.8 g上述前驱体放入H2等离子体的石英管反应器中,在一定的H2流速下(5、150 mL/min),调节输出电压升至30 V,停留10 min,然后采用2种速率升至输出终压(40~70 V)。第一种是每隔 5 V 停留2 min,升至输出终压(缓慢升高电压);第二种是从30 V直接升至输出终压,中间不停留(快速升高电压)。升至输出终压后需保持一定还原时间(60~120 min),待还原结束后,所制备Ni2P催化剂在25 ℃下经10%体积分数H2S/Ar钝化2 h,制得Ni2P催化剂样品(部分制备的Ni2P催化剂需经洗涤过程,一份为水洗3次,一份为氨水洗涤3次后再用水洗涤3次,60 ℃烘干6 h后得到终产物)。
以次磷酸为磷源,采用H2等离子体还原法在还原终压50 V、还原时间60 min、H2还原气速150 mL/min、快速升高电压的条件下,分别制备的未经水洗、经水洗和经氨水洗涤的催化剂样品记为Ni2P(O)、Ni2P(W)和Ni2P(A)。
作为参比,以硝酸镍为镍源,磷酸氢二铵为磷源,分别采用H2等离子体法(PR)[13]和程序升温还原法(TPR)[14]制备Ni2P催化剂,所制备的催化剂样品记为Ni2P(PR)和Ni2P(TPR)。H2等离子体法制备Ni2P催化剂的条件:还原终压为70 V,H2流速为150 mL/min,还原时间为120 min。程序升温还原法制备Ni2P催化剂的条件:以2 ℃/min升温速率从25 ℃升温至400 ℃并保持10 min,然后以 1 ℃/min 升温速率升到500 ℃,保持2 h,H2流速为150 mL/min。所制备Ni2P催化剂在25 ℃下经10%体积分数H2S/Ar钝化2 h。
1.3 Ni2P催化剂的表征
1.3.1 X射线衍射分析
采用Rigaku D/Max 2400型X射线衍射仪(XRD)对Ni2P催化剂样品的晶相进行表征,CuKα辐射源,Ni滤波,管电压为40 kV,管电流为100 mA。
1.3.2 电感耦合等离子体发射光谱分析
采用美国Perkin Elmer公司生产的Optima 2000DV型电感耦合等离子发射光谱仪对制备的部分Ni2P催化剂样品进行元素分析,并计算了组成催化剂的元素摩尔比。
1.3.3 傅里叶红外光谱分析
采用Equinox 55型傅里叶红外光谱仪测定Ni2P催化剂样品的吡啶吸附红外光谱,采用KBr稀释样品压片,仪器的分辨率为4 cm-1,扫描次数64次。检测采用透射法,检测范围400~2000 cm-1。
1.4 Ni2P催化HDS反应活性评价
采用固定床反应器进行Ni2P催化剂的HDS催化性能评价。催化剂装填在内径为8 mm的不锈钢中。称取0.2 g Ni2P催化剂,装入固定床反应器中,床层两端用石英砂填装。用高压计量泵将反应原料(质量分数为0.8%DBT的十氢萘溶液)注入到固定床反应器中。反应条件为:总压4.0 MPa,液时空速(LHSV)为26.25 h-1,氢/油体积比750,反应温度300~360 ℃。采用HP-6890+型气相色谱仪测定原料和反应产物的组成。色谱柱为Agilent公司HP-5型毛细柱,固定相为5%二苯基-95%二甲基聚硅氧烷(质量分数)。
DBT的转化率(x)可以采用公式(1)来计算。
(1)
其中,cDBT0为原料中DBT的摩尔浓度,mol/L;cDBT为产物中DBT的摩尔浓度,mol/L。
HDS反应各个产物的选择性(s)可以采用公式(2)来计算。
(2)
其中,ci为各产物的摩尔浓度(i为CHB、BP、HH-DBT、THDBT),mol/L。
2 结果与讨论
2.1 制备条件对以次磷酸为前体制备的Ni2P催化剂反应性能的影响
2.1.1 还原终压的影响
图1为以次磷酸为磷源的催化剂前体在H2流速150 mL/min、缓慢升高电压条件下制备的体相Ni2P催化剂的XRD谱图。如图1(a)所示,在还原终压为70 V时,制备的催化剂样品除含有Ni2P的特征峰外,还出现Ni(PO3)2(PDF 28-0708)的特征衍射峰。文献[15-16]认为,次磷酸盐在还原过程中会发生如式(3)所示的歧化反应,生成无定形的H3PO4(这与所制备的催化剂样品水洗后水洗液呈酸性结果相一致),H3PO4在高温条件下不稳定失水后生成酸酐P4O10,Ni2P在水蒸气和P4O10存在的条件下会发生如式(4)所示的反应,进而生成高温下难以被还原的Ni(PO3)2[17-18]。因此,以次磷酸为磷源时,较高还原终压(70 V)不利于纯Ni2P晶相的生成。
12Ni(H2PO2)2+35H2→
6Ni2P+14PH3+32H2O+4H3PO4
(3)
2Ni2P+2P4O10+4H2O→
4Ni(PO3)2+2PH3+H2
(4)
对介质阻挡等离子体放电过程,还原终压表示输入功率的大小[19],较高的输入功率会增加活性氢物种的数量并促进次磷酸盐的歧化分解和还原过程。为避免次磷酸盐过快地歧化分解,实验将还原终压降低到40~60 V,此时Ni(PO3)2晶相消失,但出现归属于Ni2P4O12·10H2O(PDF 31-0909)的特征峰。随着还原终压的逐渐降低,Ni2P4O12·10H2O的特征峰强度逐渐增加。这是由于气速较快时,歧化生成的PH3很容易随着气体被带出,而较低的温度不足以使生成的水分全部移除,导致Ni2+、PO43-和OH-之间会发生相互作用,进而生成Ni2P4O12·10H2O[20-21]。增加还原时间并不能将Ni2P4O12·10H2O特征峰消除,说明在此条件下Ni2P4O12·10H2O稳定存在。实验表明,在40 V时Ni2P4O12·10H2O含量最高,说明Ni2P4O12·10H2O可能是在低还原终压条件下生成的,且生成后在低于60 V的条件下稳定存在。

图1 不同还原终压和还原时间条件下制备的Ni2P催化剂的XRD谱图Fig.1 XRD patterns of Ni2P catalysts prepared with different reduction voltages and reduction timeReduction voltage/V: (a) 70; (b) 60; (c) 50; (d) 40 Ni2P; Ni2P4O12·10H2O; Ni(PO3)2
2.1.2 还原时间的影响
由图1还可以看出,以次磷酸为磷源的催化剂前体在H2流速150 mL/min、缓慢升高电压条件下制备体相Ni2P时,在还原终压40、50、60、70 V下所制备的催化剂样品均表现出不纯Ni2P的特征衍射峰。且随着还原时间的延长,催化剂样品并没有发生太大变化。
2.1.3 升电压速率的影响
为证实Ni2P4O12·10H2O可能是在较低还原终压(40 V)下生成这一结论,在H2流速为150 mL/min条件下,分别在还原终压40、50 V时通过快速升高电压方法制备了Ni2P催化剂样品。通过快速升高电压,减少反应体系在较低电压下存在的时间,避免还原过程中Ni2+、PO43-和OH-相互作用生成Ni2P4O12·10H2O。图2为不同升电压速率条件下制备的Ni2P催化剂的XRD谱图。可以看出,当还原终压为40 V时,快速升高电压(即不在35 V停留),仍然会出现Ni2P4O12·10H2O晶相。而当还原终压为50 V时,快速升高电压,基本可以制备出纯的Ni2P晶相;且相比终压为40 V的快速升高电压过程来说,Ni2P4O12·10H2O晶相基本消失;说明还原终压为50 V时,可以基本移除生成的水分,更加有利于制备纯的Ni2P晶相。因此,改变电压的升高速率对催化剂前体的还原有着直接的影响,快速升高电压有利于生成纯相的Ni2P;而较慢速率升高电压会导致生成的Ni2P晶相不纯。结合2.1.1节可知,制备纯相Ni2P的最低适宜还原终压为50 V,且制备时应采取快速升高电压的方式。
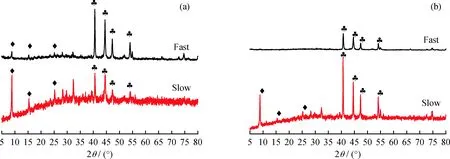
图2 不同升电压速率下制备的Ni2P催化剂的XRD谱图Fig.2 XRD patterns of Ni2P catalysts prepared at different voltage ratesReduction voltage/V: (a) 40; (b) 50 Ni2P; Ni2P4O12·10H2O
2.1.4 还原气速的影响
图3为以次磷酸为前体,在H2流速分别为5、150 mL/min下快速升高电压方法制备的体相Ni2P催化剂的XRD谱图,从而考察H2流速对制备Ni2P催化剂的影响。可以看出,在H2流速为5 mL/min、还原终压为45 V条件下能够合成出纯相的Ni2P。然而,相同条件下,将还原终压升高到50 V时出现对应Ni(PO3)2的特征衍射峰。这可能是由于次磷酸在还原终压45 V下歧化分解产生的PH3与镍源生成Ni2P;进一步升高电压,歧化副产物H3PO4脱水生成的酸酐P4O10又与Ni2P生成Ni(PO3)2。上述结果说明,在H2流速5 mL/min、还原终压45 V下制备的Ni2P上存在无定形的磷物种,且还原终压50 V以上可能会促使其脱水生成P4O10酸酐,并与Ni2P反应生成Ni(PO3)2。将H2流速提高到 150 mL/min 时,在还原终压50 V条件下可制备出纯相的Ni2P。
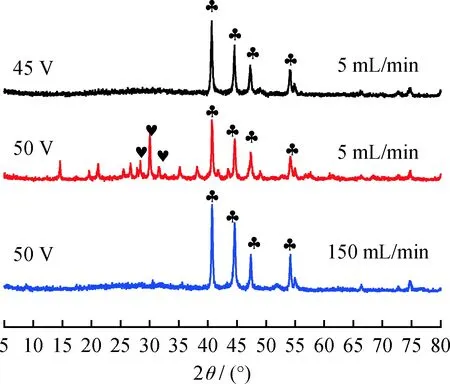
图3 不同H2流速下制备的Ni2P催化剂的XRD谱图Fig.3 XRD patterns of Ni2P catalysts preparedat different H2 flow rates Ni2P; Ni(PO3)2
上述结果说明,以次磷酸为磷源,通过H2等离子体法还原制备体相Ni2P时,还原终压、升电压速率和H2流速均会对Ni2P的生成产生影响。结合以上分析,以次磷酸为磷源制备Ni2P催化剂时,H2流速150 mL/min、快速升高电压至50 V的还原条件为最佳制备条件。
2.2 以磷酸氢二铵为前体制备的Ni2P催化剂的晶相分析
图4为以磷酸氢二铵为前体分别经过低温H2等离子体和程序程序升温还原法制备的Ni2P(PR)和Ni2P(TPR)催化剂的XRD谱图。由于磷酸氢二铵中+5价磷不发生歧化反应,反应需较高的电压来还原P—O键[13],因此低温H2等离子体法[13]采用还原终压70 V、H2流速150 mL/min、还原时间120 min条件制备出Ni2P晶相。对比图1(a),以次磷酸为磷源在该条件下制备的催化剂出现Ni(PO3)2杂峰,以磷酸二氢胺为磷源制备的Ni2P催化剂样品中并未发现其他杂晶的存在,说明Ni2P纯度很高,基本无杂质。上述结果表明,在以次磷酸为前体制备催化剂时,Ni2P4O12·10H2O、Ni(PO3)2和H3PO4等含磷杂质是在歧化过程而非还原过程中形成的。这是因为磷酸二氢胺还原制备Ni2P过程中无杂质生成。
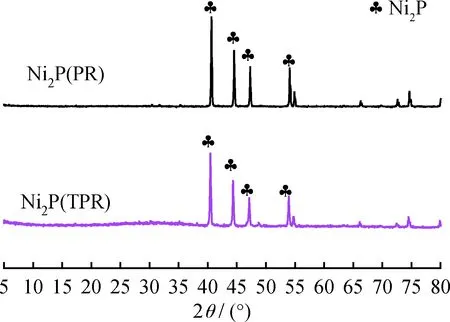
图4 Ni2P(PR)和Ni2P(TPR)催化剂的XRD谱图Fig.4 XRD patterns of Ni2P (PR) and Ni2P (TPR) catalysts
2.3 Ni2P催化剂的HDS反应性能评价及其反应后的催化剂表征结果
考察了Ni2P(O)、Ni2P(W)、Ni2P(A)、Ni2P(PR)和Ni2P(TPR)催化剂催化DBT的HDS反应的转化率随温度的变化,结果如图5所示。由图5可以看出:Ni2P(A)和Ni2P(PR)催化剂活性最高;DBT在 300 ℃ 的转化率分别在10%和20%左右,360 ℃ 时转化率能达到接近100%;且随着温度的升高,Ni2P(A)的活性逐渐高于Ni2P(PR)。Ni2P(W)催化时,DBT在300 ℃时的转化率为10%左右,360 ℃时转化率能达到90%左右。Ni2P(O)和Ni2P(TPR)催化剂活性最低,DBT在300 ℃的转化率为10%,360 ℃时分别能达到60%和50%。
前期研究结果证实,H2等离子体法制备的催化剂性能要明显优于程序升温还原法制备的催化剂性能[22]。这与Ni2P(TPR)表现出较低活性的结果相一致。以次磷酸为磷源制备的催化剂活性与后处理条件密切相关,未水洗样品(Ni2P(O))活性低于水洗样品(Ni2P(W)),水洗样品活性低于氨水洗涤的样品(Ni2P(A))。这说明次磷酸歧化生成的含磷杂质会严重抑制Ni2P的HDS反应性能,水洗并不能完全除去生成的含磷杂质,而氨水洗涤可以有效去除生成的含磷杂质,使活性大幅提高。次磷酸在歧化过程中可以生成磷酸等含磷杂质[17],含磷杂质会覆盖催化剂的活性中心,导致反应过程中可接近活性中心的数目降低,抑制了Ni2P的HDS反应性能[18]。Ni2P(O)在320 ℃出现反常的低活性,甚至低于300 ℃时的活性。水洗和氨水洗涤后的样品(Ni2P(W)和Ni2P(A))并未出现相似的规律,说明320 ℃的低活性是由于可被洗掉的含磷杂质导致的。当温度高于320 ℃时,含磷杂质可能在反应条件下逐渐消失,因此340 ℃活性明显升高。

图5 不同Ni2P催化剂催化DBT的HDS反应转化率(x)随温度(T)的变化Fig.5 Relationship between the conversion rate (x) of DBTand temperature (T) in HDS reaction over different catalysts(1) Ni2P(PR); (2) Ni2P(A); (3) Ni2P(W);(4) Ni2P(TPR); (5) Ni2P(O)Reaction conditions: m(Ni2P)=0.2 g; p=4.0 MPa;LHSV=26.25 h-1; v(H2)=75 mL/min; v(Raw material)=0.1 mL/min
图6为HDS反应前后5种Ni2P催化剂的XRD谱图。从图6可以发现,反应前后Ni2P(PR)和Ni2P(TPR)的衍射峰基本一致,均为标准Ni2P特征峰。而Ni2P(O)和Ni2P(W)在反应后均出现Ni(PO3)2的特征衍射峰,说明水洗后仍有少量含磷物种存在于催化剂上,在高温高压时与Ni2P反应生成Ni(PO3)2。相比反应前的Ni2P,反应后Ni2P的特征衍射峰的强度明显增强,可能是在高温反应的过程中,含磷物种会促进Ni2P晶粒团聚,导致其峰强度增强[1]。而Ni2P(A)催化剂在反应前后均为标准Ni2P特征峰,表明氨水洗涤已将催化剂上的含磷物种去除,也解释了Ni2P(O)、Ni2P(W)活性低于Ni2P(PR)和Ni2P(A)活性的原因。由此可以看出,在催化剂合成过程中形成的磷物种是影响其催化HDS反应性能的一个重要因素。
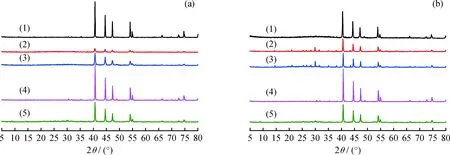
图6 HDS反应前后Ni2P催化剂的XRD谱图Fig.6 XRD patterns of the Ni2P catalysts before and after HDS reaction(a) Before HDS reaction; (b) After HDS reaction(1) Ni2P(TPR); (2) Ni2P(W); (3) Ni2P(O); (4) Ni2P(PR); (5) Ni2P(A)
图7为Ni2P(O)和Ni2P(W)样品的FT-IR谱图。由图7可知,未水洗和水洗后的Ni2P样品的FT-IR谱图基本保持一致。其中,570 cm-1和 740 cm-1处吸收峰为Ni—P的伸缩振动峰[1];510 cm-1和918 cm-1处吸收峰可归属于P—OH键伸缩振动峰[23-24];1145 cm-1和1650 cm-1处较宽的吸收峰归属于HPO42-,证明Ni2P(O)样品存在磷酸,且水洗不能将吸附的磷酸全部去除[25-26];1085 cm-1处吸收峰可归属于Ni2P中由于表面钝化产生的P=O键[27]。HDS反应后,Ni2P(W)催化剂的FT-IR光谱发生较大改变。其中,归属于Ni2P的吸收峰减小,918 cm-1和1145 cm-1处吸收峰消失,1650 cm-1处吸收峰减弱,说明磷酸含量减少;1030 cm-1处吸收峰变强,并出现可归属于磷酸盐物种的595 cm-1特征峰,表明HDS反应后Ni2P(W)催化剂上的磷物种发生了重构。这与XRD谱图出现杂晶的结果相一致。由ICP的数据结果可知,Ni2P(O)、Ni2P(W)、Ni2P(A)和反应后Ni2P(W)催化剂的Ni/P摩尔比分别为1.33、1.43、1.85和2.31。Ni2P(O)相比标准Ni2P表现出低于化学计量的Ni/P摩尔比,说明有含磷杂质存在于新鲜催化剂上。水洗和氨水洗涤后可部分除去含磷杂质,但Ni/P摩尔比仍低于Ni2P的化学计量比。在HDS反应后Ni2P(W)的Ni/P摩尔比升高至2.3,高于Ni2P的化学计量比,表明磷杂质在催化剂上进行了重构,产生了部分磷流失。

图7 Ni2P(O)和Ni2P(W)催化剂的FT-IR谱图Fig.7 FT-IR spectra of Ni2P(O) and Ni2P(W)(1) Ni2P(O); (2) Ni2P(W); (3) Ni2P(W) after reaction
在HDS反应中Ni2P具有2种不同配位的Ni原子,分别形成了2种不同的活性位点,即四面体配位的Ni(1)位和方椎体配位的Ni(2)位。直接脱硫(DDS)途径主要发生在Ni(1)位,主产物为联苯(BP);而加氢后脱硫(HYD)途径主要发生在Ni(2)位,主产物为苯基环己烷(CHB)和联环己烷(BCH)[1]。Ni2P(O)、Ni2P(W)、Ni2P(A)和Ni2P(PR)催化剂催化DBT的HDS反应选择性随反应温度的变化见图8。由图8可以看出,在这4种催化剂上BP均是主要的脱硫产物,水洗后的Ni2P样品除300 ℃时BP的选择性较低外,其表现出与Ni2P(PR)相似的产物分布规律。氨水洗后的Ni2P样品表现出与Ni2P(PR)基本一致的产物分布规律。这表明水洗并未全部溶解掉磷酸等含磷杂质,水洗后的洗涤液呈酸性,与上述Ni2P(O)和Ni2P(W)的红外谱图均出现磷酸的归属峰结果相一致;氨水可以与磷酸等酸性物种中和,因此氨水洗涤基本可以除去Ni2P催化剂上的含磷杂质,与反应前后Ni2P(A)的XRD谱图并未出现其他特征峰结果相一致。相比于其他3种催化剂,Ni2P(O)催化剂上加氢脱硫产物CHB选择性变化不大,但直接脱硫产物BP和加氢脱硫产物BCH选择性变化明显。可以看出,在320 ℃时,Ni2P(O)催化剂上BP选择性明显降低。这可能是在320 ℃时催化剂上的含磷杂质反应抑制了其直接脱硫路径。据文献[28-30]所知,催化剂的酸性是影响其HDS性能的重要因素,增加催化剂的总酸量,会提高催化剂的BCH选择性,降低BP的选择性。这也证实Ni2P(O)催化剂的含磷杂质具有酸活性中心。根据次磷酸的歧化方程及文献[15-16]报道可知,酸中心很可能为磷酸。继续升高温度,磷酸分解为P4O10,随后与部分Ni2P生成Ni(PO3)2,与HDS反应后Ni2P催化剂的XRD结果相一致。随着Ni2P催化剂表面酸性下降,BCH含量降低,BP含量升高。
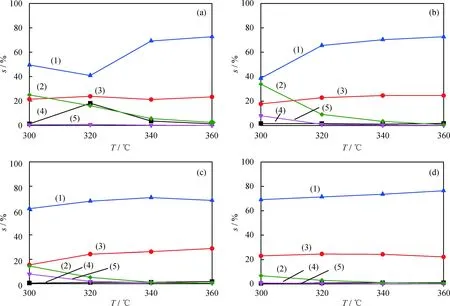
图8 DBT在不同Ni2P催化剂样品上HDS反应选择性(s)随温度(T)的变化Fig.8 Selectivity (s) of DBT on different Ni2P catalysts vs temperature (T) in HDS reactionReaction conditions: m(Ni2P)=0.2 g; p=4.0 MPa; LHSV=26.25 h-1; v(H2)=75 mL/min;v(Raw material)=0.1 mL/min(a) Ni2P(O); (b) Ni2P(W); (c) Ni2P(A); (d) Ni2P(PR)(1) BP—Biphenyl; (2) TH-DBT—Tetrahydro-dibenzothiophene; (3) CHB—Cyclohexylbenzene;(4) BCH—Bicyclohexyl; (5) HH-DBT—Hexahydro-dibenzothiophene
3 结 论
(1)次磷酸经歧化生成的磷酸和磷酸缩合生成的P4O10酸酐会导致合成Ni2P催化剂中含磷杂质的生成,进而抑制所制备的Ni2P催化剂的HDS反应性能。
(2)以次磷酸为磷源制备的Ni2P催化剂,其含磷杂质可以通过水洗部分去除,从而使水洗后Ni2P催化剂的HDS催化活性有所提高,但水洗无法全部去除Ni2P催化剂上的含磷杂质。氨水洗涤可以更有效地去除歧化过程生成的含磷杂质,使Ni2P活性大幅提高。未水洗Ni2P催化剂存在较强的酸中心,且该酸中心在340 ℃的HDS反应中会逐渐消失,HDS反应后的Ni2P催化剂会生成Ni(PO3)2晶相。
(3)Ni2P催化剂上的含磷杂质会与Ni2P反应,进而影响Ni2P晶相的稳定存在。在等离子体反应器中,以次磷酸为磷源,在H2流速150 mL/min下,通过快速升高电压至50 V可制备出较纯Ni2P催化剂,通过氨水洗涤可有效去除催化剂上的含磷杂质;以磷酸氢二铵为磷源制备催化剂会避免含磷杂质的生成,有利于制备高温高压下稳定存在的Ni2P催化剂。
