1例遗传性凝血因子Ⅶ缺陷症家系基因突变分析
2020-03-23郭跃丽孔万仲万菁蔡文品陈孟权奚经巧温州市中医院检验科浙江温州325000
郭跃丽,孔万仲,万菁,蔡文品,陈孟权,奚经巧(温州市中医院检验科,浙江温州325000)
遗传性凝血因子Ⅶ(FⅦ)缺陷症是一种罕见的常染色体遗传性出血性疾病,常见于近亲婚配的家族。大部分患者主要表现为黏膜出血,例如鼻出血、牙龈出血、瘀斑等,女性患者可表现为月经量增多[1-2]。其临床表现具有高度异质性,出血严重程度与基因突变位点、突变数量、突变类型等密切相关[3]。因此,分析F7基因突变对遗传性FⅦ缺陷症具有重要意义。本研究对1例近亲婚配的遗传性FⅦ缺陷症患者进行凝血指标检测与基因分析,初步探讨其分子发病机制。
1 资料与方法
1.1家系资料 先证者,男,48岁,汉族,浙江乐清人,因“无故反复性鼻出血”1 d于2018年3月16日就诊于我院五官科。凝血常规检查发现,血浆凝血酶原时间(PT)延长(为31.2 s),FⅦ活性(FⅦ:C)、FⅦ抗原(FⅦ:Ag)均降低(分别为4%和6%),其他凝血指标均无异常,肝、肾功能正常,平素无明显自发出血症状。其父母为姑表近亲婚配,家系其他成员均无自发出血症状。该先证者的家系图见图1。

图1 1例遗传性FⅦ缺陷症先证者的家系图
1.2健康人对照 收集150例体检健康者,其中男性86例,女性64例,年龄21~48岁,无肝、肾功能异常,无出血及血栓史,无抗凝剂用药史,女性未口服避孕药。建立本次实验室凝血表型指标的参考区间。所有研究对象均知情同意。
1.3标本采集与处理 采集研究对象空腹静脉血标本2.7 mL,用0.109 mol/L枸橼酸钠溶液抗凝。标本3 000 r/min离心15 min后,取上层乏血小板血浆用于各凝血指标的检测,2 h内完成;下层血细胞于-40 ℃冻存,待用于基因组DNA的抽提。
1.4凝血指标检测 PT、凝血活酶时间(APTT)的检测采用一期凝固法;纤维蛋白原(Fib)的检测采用Clause法;FⅦ:C、凝血因子Ⅹ活性(FⅩ:C)、凝血因子Ⅴ活性(FⅤ:C)和凝血因子Ⅱ活性(FⅡ:C)均采用基于PT的一期凝固法检测。以上均采用Stago STA-R全自动血凝仪及其原装配套试剂(法国STAGO公司)严格按照试剂说明书完成检测。
1.5外周血基因组DNA提取 选用酚-氯仿法提取先证者及其家系成员的外周血细胞基因组DNA,并用DU800核酸蛋白分析仪检测所提取基因组DNA的浓度和纯度。所有步骤均按照酚-氯仿法基因组DNA提取试剂盒说明书进行,基因组DNA提取试剂盒由北京天根生物公司提供。
1.6引物及PCR扩增 根据F7基因序列(GenBank J02933),用Primer Premier 5.0软件分别设计11对引物以覆盖F7基因的所有外显子、5′和3′非翻译区序列、侧翼,引物序列及PCR扩增条件参见文献[3]。引物由上海桑尼生物公司合成。
1.7PCR产物测序 PCR产物割胶后送上海桑尼生物工程公司纯化后用ABI3730XL型测序仪(美国Applera公司)测序。通过Chromas软件与美国NCBI 基因库所公布的F7基因序列(GenBank GI180333和J02933序列)以及F7突变数据库(http://europium.csc.mrc.ac.uk)在Blaster软件上(http:http://www.ncbi.nlm.nih.gov/blast)进行比对,查找基因突变的位点,发现突变位点后,再利用反向测序予以证实。明确先证者基因突变的位点后,利用PCR扩增其他家系成员的相应突变位点区域并进行测序分析。
1.8生物信息学技术分析 采用ClustalW软件分析氨基酸突变位点保守性,采用PolyPhen-2、SIFT软件分析氨基酸突变前后生物信息学特性,采用Swiss-pdb Viewer软件分析FⅦ蛋白模型野生型和突变型局部空间构型及分子间作用力的变化。
2 结果
2.1先证者及其家系成员凝血指标水平 先证者PT明显延长;FⅦ:C和FⅦ:Ag较健康人对照降低。先证者母亲、父亲、大姐、二姐和哥哥PT均稍延长;FⅦ:C和FⅦ:Ag均下降至健康人对照的一半左右,家系成员的其他凝血指标均无异常(表1)。

表1 先证者及其家系成员主要凝血指标检测结果
2.2先证者及其家系成员基因分析 先证者F7基因(GenBank GI180333)第8外显子存在c.1224 T>G纯合错义突变导致p.His348Gln,其母亲、父亲、大姐、二姐和哥哥均存在c.1224 T>G杂合错义突变,其外甥女为野生型(图2)。
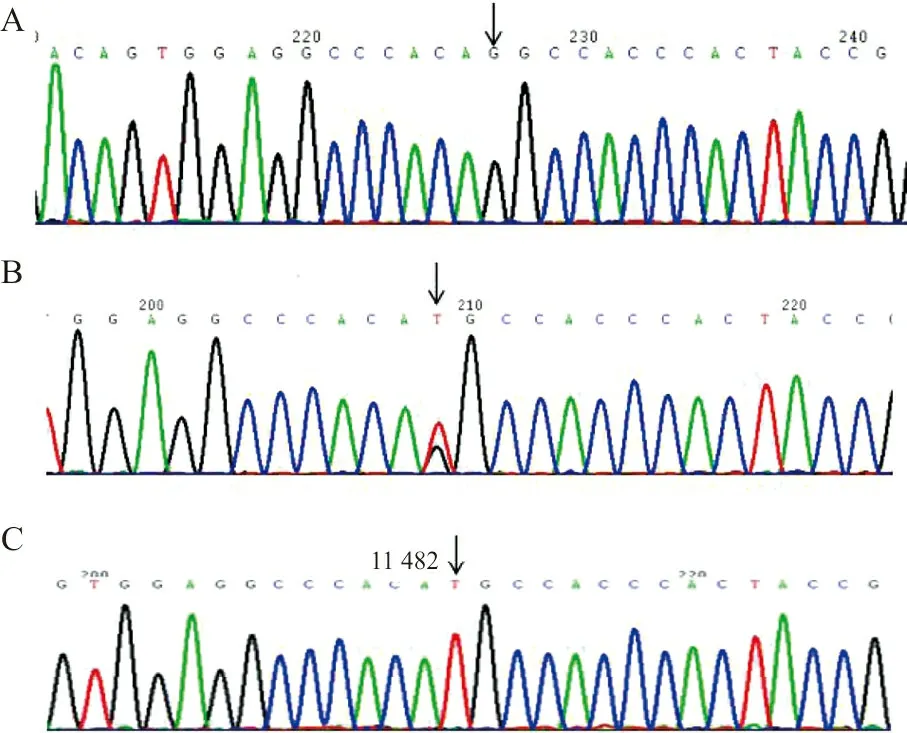
注:A,先证者c.1224T>G纯合子突变;B,母亲c.1224T>G杂合子突变;C,正常序列;箭头示突变位置。
图2遗传性FⅦ缺陷症家系第8号外显子测序结果
2.3生物信息学技术分析 用ClustalⅩ-2.1-win软件对人类和 NCBI 数据库中提供的其他9种同源性物种F7基因氨基酸序列进行多重比对。图3中箭头所指为FⅦ的348位氨基酸His,目标线全部为H,表明该氨基酸在同源物种中具有高度保守的特性。

图3 不同物种间FⅦ的His348的氨基酸序列比对
分别用PolyPhen-2和SIFT在线生物信息学软件对FⅦ突变后蛋白质通过评分预测功能:His348Gln的PolyPhen-2预测结果为1.000,提示“可能是有害的”;SIFT预测结果为0.32,提示“影响蛋白质功能”。用Swiss-Pdb Viewer version软件对FⅦ His348Gln突变前后的蛋白质结构进行模型分析:野生型蛋白质的分子模型结构中His348与Thr359和Gly360之间存在5条氢链,当亲水性的Gln348取代碱性的His348时芳香环消失,Gln348与Thr359之间少了1条氢链。见图4。
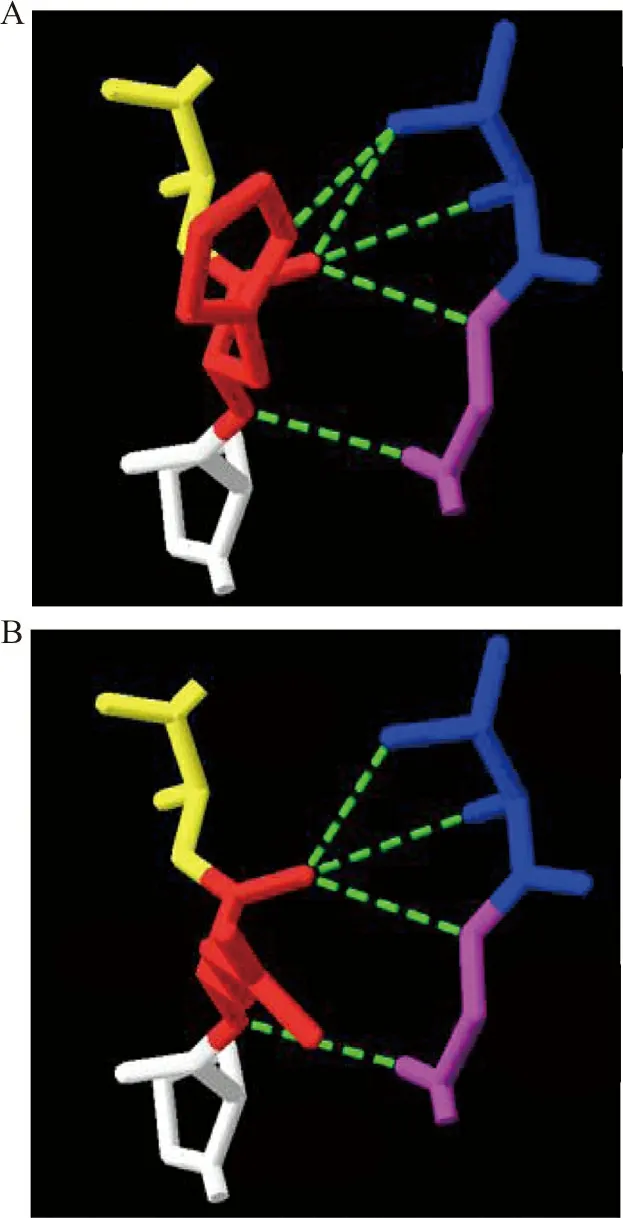
注:A,野生型;B,His348Gln突变型;绿色虚线表示氢键。
3 讨论
遗传性FⅦ缺陷症是一种常染色体隐性遗传性出血性疾病,其临床表现与血浆FⅦ:C无明显相关性,重症患者多为影响基因结构和功能的纯合或者复合杂合突变所致,其FⅦ:C通常低于2%;而单一杂合突变患者的FⅦ:C一般不低于30%,几乎无临床症状[4]。大约有1/3的FⅦ缺陷症患者可终身无出血表现,这些患者往往是在家族史研究或者止血试验筛查时被发现[5]。本研究家系中的先证者凝血常规筛查发现PT延长、FⅦ:C和FⅦ:Ag减低,诊断为FⅦ缺陷症。通过先证者和家系成员的基因分析发现:先证者存在F7基因c.1224T>G纯合子错义突变,分别遗传自携带c.1224 T>G杂合错义突变的父亲和母亲。先证者F7基因第8号外显子存在纯合错义突变c.11482 T>G,导致p.His348Gln。家系中母亲、父亲、大姐、二姐和哥哥均存在c.1224 T>G杂合错义突变,其FⅦ:C和FⅦ:Ag均约下降至健康人对照的一半,与国内外相关报道[6-7]一致。
国际上关于F7基因His348Gln突变位点的第1个病例由日本Katsumi教授报道[8],在对该突变位点进行体外表达研究发现,该突变可改变FⅦ的空间结构,导致其分泌障碍,致使血浆中的FⅦ:C及FⅦ:Ag均降低,表现为交叉反应物质阴性(CRM-),表明His348Gln纯合突变会影响FⅦ的分泌功能。Ding等[9]研究了其致病机制,亦证实His408(348)Gln 双杂合突变影响FⅦ的合成。本研究家系发现的His348Gln突变位点用Swiss-Pdb Viewer version 软件对突变前后的蛋白质结构进行模型分析,结果显示:野生型蛋白质的分子模型结构中His348与Thr359和Gly360之间存在5条氢链,当亲水性的Gln348取代碱性的His348时芳香环消失,Gln348与Thr359之间缺失了1条氢链。FⅦa与组织因子(TF)结合的活化位点在Met306[10],将该位点的Met进行氨基酸替换后,可打破原有的正常氢链结构,从而阻断TF对FⅦ的活化过程。本研究报道的His348位于Met306附近,蛋白质结构分析显示突变后可导致原有氢链数量的减少。因此我们推测该突变可能也会影响TF对FⅦ的活化过程,是否由于TF-FⅦa复合物在不同情况下存在不同的生理活性,从而使F7基因即使具有相同的突变位点,但若还有其他影响TF-FⅦa复合物形成的因素存在的情况下,仍可导致不同的临床表现。
