大肠杆菌cpxR 和hns 双基因缺失株的构建
2020-03-13胡慧慧孙亚伟李文娅邝启红孙华润胡功政河南农业大学牧医工程学院河南郑州450046河南科技学院动物科技学院河南新乡45300
胡慧慧,孙亚伟,李文娅,邝启红,孙华润,吴 华,胡功政,苑 丽∗ (.河南农业大学 牧医工程学院,河南 郑州450046;.河南科技学院 动物科技学院,河南 新乡45300)
在肠杆菌科细菌中,质粒接合作用是水平基因传播(horizontal gene transfer,HGT)最主要的方式之一。参与质粒接合作用的调控蛋白有很多,其中H-NS核蛋白[1]Cpx AR/Arc AB 双组分信号转导系统(two-component signal transduction system,TCS)[2]发挥了很重要的作用。H-NS 核蛋白是肠杆菌科细菌中一种核酸样DNA 结合蛋白,能识别和选择性沉默外源性基因,从而可有效避免细菌因获得和表达过多外源性基因而耗费大量的能量[3-5]。双组分信号转导系统是革兰阴性菌体内常见的调控元件之一,能调控多种基因的表达,是目前极具吸引力的对原核生物有高度选择性的抗菌作用新靶标[6],其中Cpx AR 是肠杆菌科细菌中常见的双组分信号转导系统[2]。
采用 Red 同源重组[7-11],对大肠杆菌ATCC25922中cpx R和hns基因进行敲除,构建单/双基因缺失株及它们的回补菌株,为研究H-NS及Cpx AR 互作调控IncFⅡ质粒接合作用的分子机制奠定基础。
1 材料与方法
1.1 菌株与质粒菌株:大肠埃希菌ATCC®25922,购自中国普通微生物菌种保存中心,由本实验室保存。质粒:p KD4,两侧含有FRT 位点的卡那霉素抗性(kanr)基因,是基因敲除的辅助性质粒。p KD46,同源重组的协助质粒,含有温度敏感型复制子和氨苄西林(Amp)抗性,经L-阿拉伯糖诱导后能够表达Gam、Beta和Exo等3种同源重组酶。p CP20质粒,具有氯霉素和Amp 抗性,用于消除FRT 位点的kanr基因[12];pBAD/His A 质粒,具有Amp抗性,主要用于构建cpx R和hns的过表达载体。
1.2 培养基及主要试剂LB肉汤、LB琼脂均购于北京路桥技术有限公司;SOC 购于北京索莱宝生物科技有限公司;质粒小提试剂盒和DNA 胶回收试剂盒购于美国OMEGA 公司;高保真DNA 聚合酶,XholⅠ和Hind Ⅲ内切酶,T4DNA 连接酶、PCR Mix、DpnⅠ酶等均购于Ta KaRa 公司。BM2000 DNA Marker为Bio Med公司产品;Trans2K DNA Marker和Trans5K DNA Marker购于北京全式金生物技术有限公司。氨苄西林钠、卡那霉素均购于北京索莱宝科技有限公司。
1.3 菌株的复苏取甘油保存的菌种,按1∶100比例无菌接种于5 m L 新鲜的LB 肉汤,置于37℃震荡培养12~16 h,然后用接种环无菌挑取一环菌液划线接种于LB 琼脂平板上,倒置于37℃温箱中培养12~16 h,后钓取单个菌落接种于LB 肉汤,37℃震荡培养12 h后备用。
1.4 质粒提取p KD4、p KD46、pCP20、pBAD/His A 等质粒的提取按照美国OMEGA 公司的质粒小量提取试剂盒说明书进行,将收集的液体置于-20℃保存。
1.5 引物设计
1.5.1 缺失株构建所用引物 根据GenBank公布的cp x R和hns的核苷酸序列,应用Primer 5.0分析软件设计6对引物(表1)。其中LCR-F/LCR-R和LHN-F/LHN-R 为敲除引物,用于PCR 扩增同源重组的线性打靶片段;CR-F/CR-R 和HN-F/HN-R 为cp x R和hns敲除鉴定引物,用于检测cpx R/hns缺失突变株;KF/KR 用于检测插入的kanr基因;引物组合CR-F/KR、KF/CR-R、HN-F/KR、KF/HN-R 用于验证kanr基因的插入位置[13]。
1.5.2 回补菌株构建所用引物 参照GenBank上已公布的cpx R和hns的全基因序列,用Primer 5.0软件设计4 对引物(表1)。其中cp x R-1-F/cp x R-1-R、hns-1-F/hns-1-R,用于扩增包含cpxR 和hns的全基因长片段,cpx R-F/R 或hns-F/R 是带有双酶切位点的cpx R、hns完整的开放阅读框(open reading frame,ORF)。
1.6 cpxR 和hns 缺失株的构建
1.6.1 线性打靶片段的制备及鉴定 以p KD4为模板,分别用引物LCR-F/LCR-R 和LHN-F/LHN-R扩增线性打靶片段(中间含kanr基因,两侧包含cpx R/hns同 源 臂 的PCR 产 物)。所 得PCR 产 物 用OMEGA 公司的DNA 胶回收试剂盒进行纯化,纯化后的胶回收产物用DpnⅠ酶消化后,分别加入适量的DpnⅠ酶和10×T Buffer,37℃过夜消化,用胶回收试剂盒纯化,测序鉴定。
1.6.2cpx R和hns基因缺失 将p KD46质粒电转化至新制备的大肠杆菌ATCC25922 感受态细胞中,在含有适量Amp 的LB 平板上筛选,获得ATCC25922/p KD46菌株。
ATCC25922/p KD46 菌株经L-阿拉伯糖诱导后作为受体,将1.6.1获得的线性打靶片段电击转化入受体中,在含有适量Kan的LB 平板上筛选转化子。用CR-F/CR-R 或HN-F/HN-R、CR-F/KR或HN-F/KR、KF/CR-R 或KF/HN-R、KF/KR 4对引物分别进行PCR 扩增验证,PCR 产物送擎科公司测序鉴定。
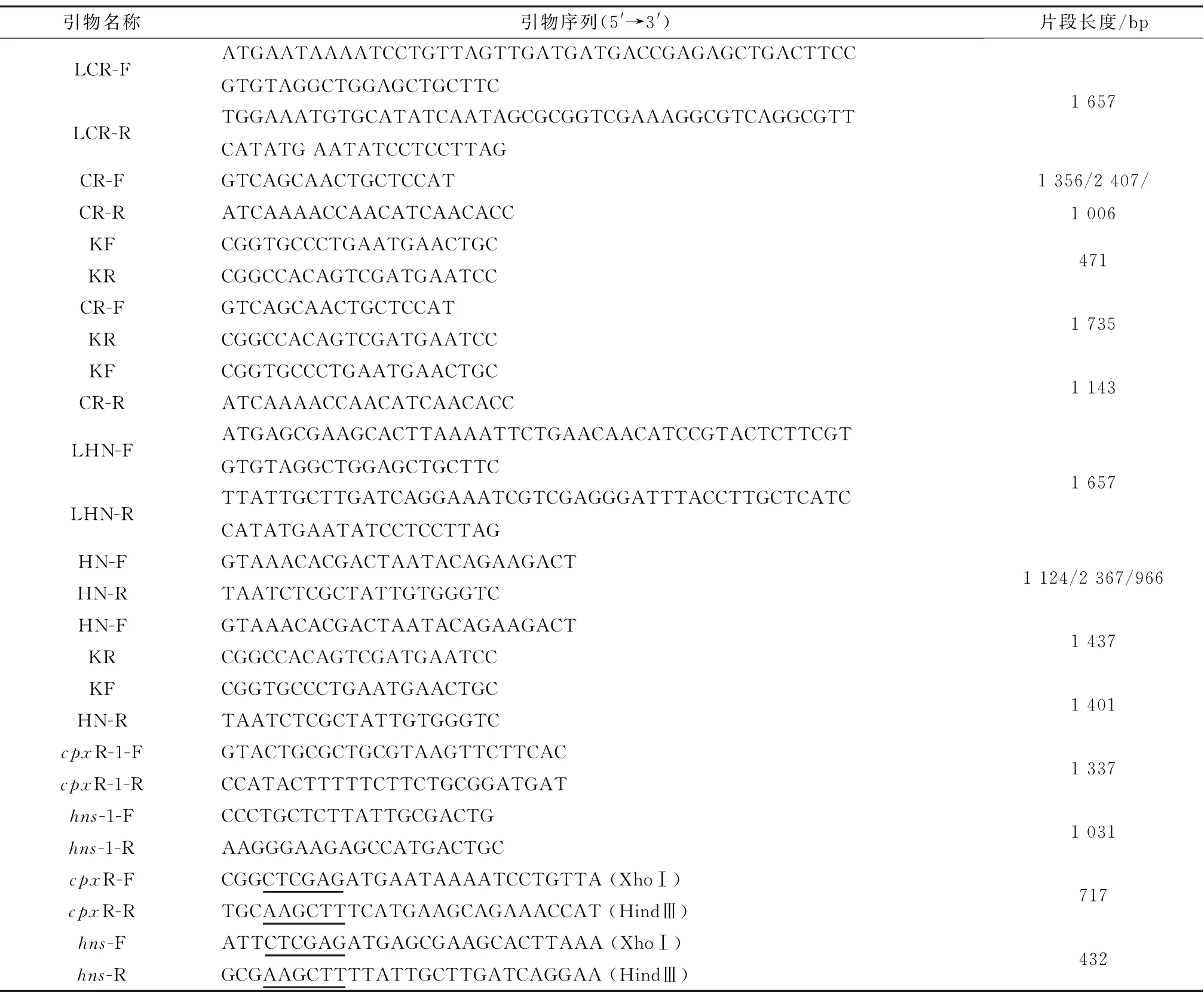
表1 cpx R 和hns 敲除所用引物
1.6.3 p KD46和kanr基因的消除 挑取鉴定成功的阳性转化子接种LB 平板,43℃培养6 h,将培养物接种于含Amp的LB 平板,37℃培养18 h观察是否有细菌生长。随后,将除去p KD46的阳性转化子接种于含适量卡那霉素的LB上培养。
将p CP20 电转入阳性转化子中,然后接种在LB(含氯霉素和Amp)平板上,30℃过夜培养后,挑取单个菌落接种于LB 肉汤,43℃培养6 h,将菌液分别接种于含Amp/Kan 的LB 平板,37℃培养18 h,如无细菌生长,则上述菌液即为目标重组菌,分别用检测引物CR-F/CR-R、HN-F/HN-R 对重组菌进行PCR 扩增验证,并测序鉴定,确认为缺失株的分别命名为△cp x R和△hns。
1.7 构建cpx R 和hns 双基因缺失株以菌株△cp x R为研究对象,利用Red同源重组技术将两侧包含hns同源臂的线性打靶片段替代△cpx R菌中的hns基因,构建cp x R和hns的双基因缺失株(标记为△cpx R△hns:kanr),用检测引物CR-F/CR-R、HN-F/HN-R、HN-F/KR、KF/HN-R、KF/KR 分别进行PCR 扩增验证,并测序鉴定。
1.8 回补菌株的构建以大肠杆菌ATCC25922的DNA 为模板,用引物cpx R-1-F/cpx R-1-R 和hns-1-F/hns-1-R 分别扩增包含cpx R和hns的全基因长片段。再以PCR 扩增产物为模板,用cpx R-F/cpx R-R 或hns-F/hns-R 扩增出含有酶切位点的cpxR、hns完整ORF。PCR 产物经胶回收纯化后,与p BAD/His A 质粒同时用XholⅠ和Hind Ⅲ进行双酶切,酶切产物经纯化试剂盒纯化后,用T4DNA Ligase 4℃过夜连接,连接产物用化学转化法转入DH5α感受态细胞。用引物cpx R-F/cpx R-R、hns-F/hns-R 进行PCR 扩增验证,提取重组质粒用XhoⅠ和HindⅢ双酶切鉴定,并送擎科公司进行测序比对,确证后分别标记为cp x R-pBAD/His A、hnspBAD/His A 重组质粒。
将构建成功的cpx R-p BAD/His A、hns-p BAD/His A 重组质粒分别电击转化入感受态细胞△cpx R、△hns和△cpx R△hns:kanr中,在 含 适 量Amp的LB平板上筛选阳性转化子,即为回补菌株,分 别 命 名 为△cpxR/pcpxR、△hns/phns、△cpxR△hns/pcpxR和△cpxR△hns/phns。
2 结果
2.1 cpxR 和hns 单基因缺失株的鉴定cpx R和hns单基因缺失株在含有Kan的LB平板上长成边缘光滑、湿润、有光泽、白色略透明的单个小菌落。分别用CR-F/CR-R 或HN-F/HN-R、CR-F/KR 或HN-F/KR、KF/CR-R 或KF/HN-R、KF/KR 4 对引物进行PCR 扩增验证时,均出现预期大小的目的片段(图1,2),测序结果显示,cpx R和hns基因均已被线性打靶片段代替,分别标记为△cpx R:kanr和△hns:kanr。
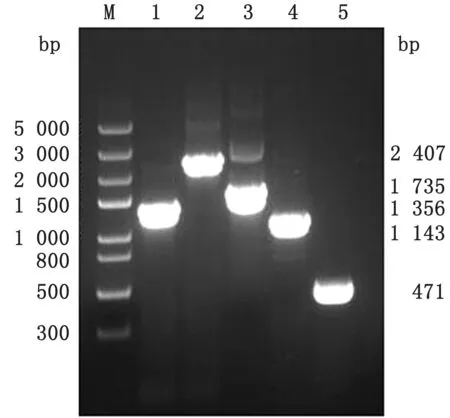
图1 cpx R 基因Red 同源重组PCR 鉴定电泳图 M.Trans5000 DNA Marker;1.CR-F/CR-R 引 物 扩 增ATCC25922中cpx R 的PCR 产 物;2.CR-F/CR-R 引物扩增△cpx R:kan r中打靶片段替代cpx R 后的片段;3.CR-F/KF 引物扩增kan r基因插入位点上游片段;4.KR/CR-R 引物扩增kan r基因插入位点下游片段;5.用KF/KR 引物扩增kan r基因内的片段
2.2 消除kan r的鉴定以△cpx R:kanr、△hns:kanr、△cpx R和△hns为模板,CR-F/CR-R 和HNF/HN-R 为引物,PCR 扩增cpx R和hns基因,结果均得到与预期一致的目的条带(图3,4)。测序结果显示kanr已被成功消除,仅残留长为83 bp的FRT位点,说明利用此方法已成功将△cp x R:kanr和△hns:kanr中的kanr基因消除,构建出△cp x R和△hns的单基因缺失株。
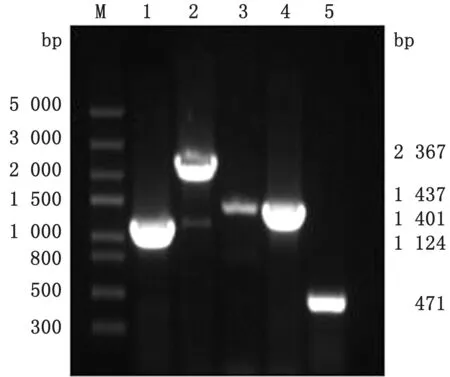
图2 hns 基因Red 同源重组PCR 鉴定电泳图 M.Trans5000 DNA Marker;1.HN-F/HN-R 引 物 扩 增ATCC25922中hns 的PCR 产 物;2.HN-F/HN-R 引物扩增△hns:kan r中打靶片段替代hns 后的片段;3.HN-F/KF 引物扩增kan r基因插入位点上游片段;4.KR/HN-R 引物扩增kan r基因插入位点下游片段;5.KF/KR 引物扩增kan r基因内的片段
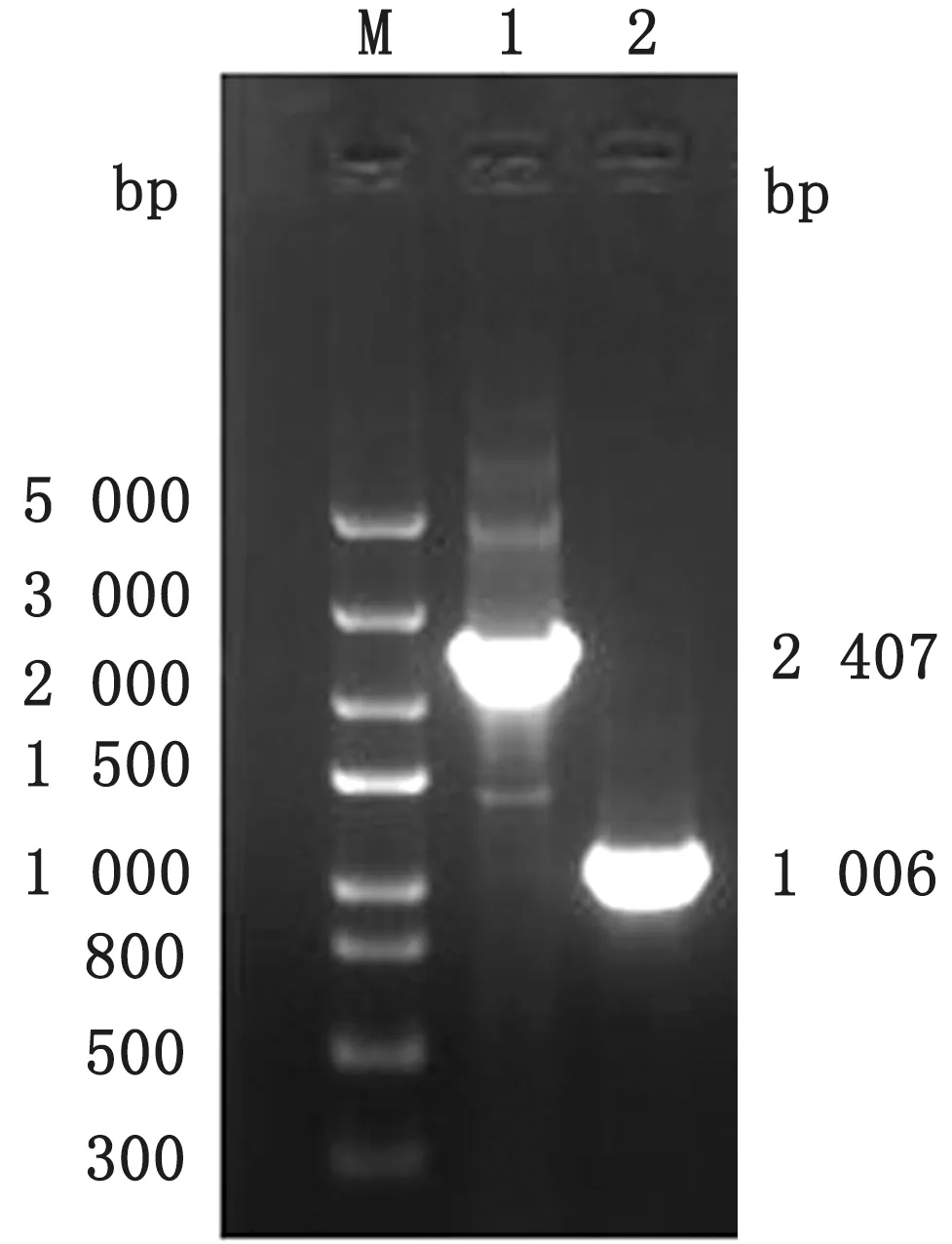
图3 消除△cpx R:kan r 株的kan r 基因电泳图 M.Trans5000 DNA Marker;1~2.以△cp x R:kan r 和△cpx R 为模板用引物CR-F/CR-R 扩增cp x R 的电泳条带
2.3 cpxR 和hns 双基因缺失株的鉴定以构建的双基因缺失株△cp x R△hns:kanr为模板,用CR-F/CR-R、HN-F/HN-R、HN-F/KR、KF/HN-R 和KF/KR 等5对引物分别进行PCR 扩增进行鉴定,结果扩增出预期大小的片段(图5)。
2.4 回补菌株的构建及鉴定将cpx R、hns的完全开放阅读框分别连接pBAD/His A 载体并转入感受态细胞DH5α后,提取重组质粒,经PCR 鉴定均分别扩增出cpx R和hns的完整ORF,经XhoⅠ和HindⅢ双酶切(图6)证实为cpx R-p BAD/His A 和hns-p BAD/His A 重组质粒。
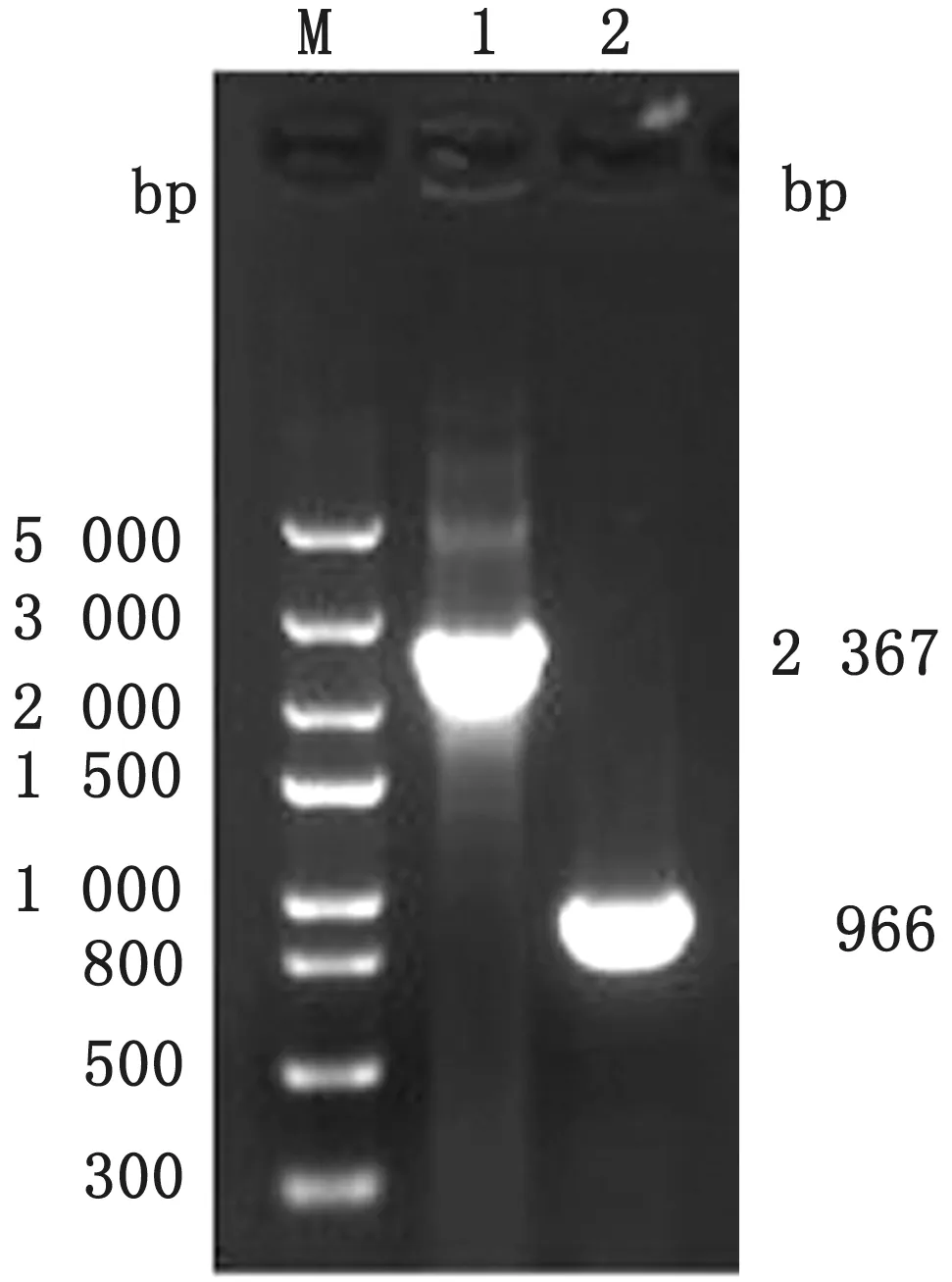
图4 消除△hns:kan r株的kan r基因电泳图 M.Trans5000 DNA Marker;1~2.以△hns:kan r和△hns 为模板用引物HN-F/HN-R 扩增hns 的电泳条带
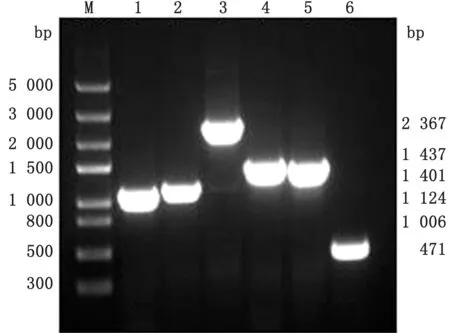
图5 △cpx R△hns:kan r双基因缺失株的鉴定 M.Trans5000 DNA Marker;1.CR-F/CR-R 引 物 扩 增△cpx R 中cpx R 基 因;2.HN-F/HN-R 引 物 扩 增△cpx R 中hns基因;3.HN-F/HN-R 扩增△cpx R△hns:kan r中打靶片段替代hns 后的片段;4.HN-F/KR 扩增△cpx R△hns:kan r中kan r基因插入位点上游片段;5.用KF/HN-R 扩增△cpx R△hns:kan r中kan r基因插入位点下游片段;6.用KF/KR 扩增kan r基因内的片段
3 讨论
质粒接合作用是耐药基因在菌属间快速水平散播的最主要方式之一,但多种因素会明显制约接合型质粒接合转移频率的高低。制约因素可总结为下列2种:一为外部因素,包括供体菌细胞浓度、细胞生长速度、外界能量供给、抗菌药选择性压力等[14];另一个为内部因素,主要包括质粒转移基因(transfer,tra)的表达[15]和菌体内各种调控蛋白[16]的调控,其中tra基因正常表达是质粒发生接合转移的基础,而调控蛋白主要通过直接或间接抑制或激活tra基因转录来发挥作用[17],参与质粒接合作用的调控蛋白主要包括H-NS核蛋白和双组分信号转导系统Cpx AR/Arc AB等。
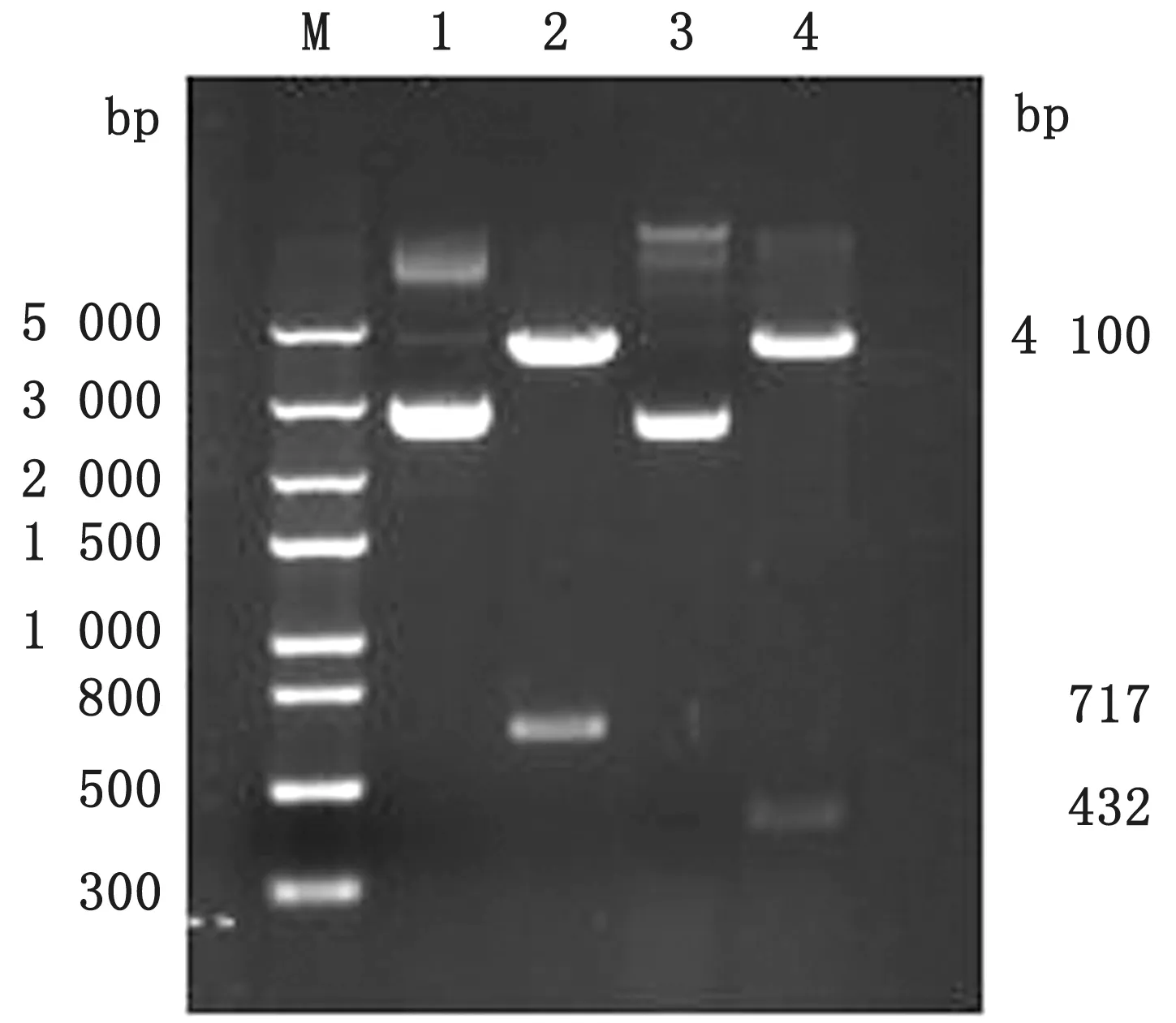
图6 cpx R 和hns 重组质粒双酶切鉴定 M.Trans5000 DNA Marker;1.cp x R-pBAD/His A 重 组 质 粒;2.cpx R-pBAD/His A重组质粒的XhoⅠ和HindⅢ双酶切条带;3.hns-pBAD/His A重组质粒;4.hns-pBAD/His A重组质粒的XhoⅠ和HindⅢ双酶切条带
本课题组在研究产CTX-M 型鸡大肠埃希菌的散播机制中发现,IncFⅡ质粒不仅是多重耐药肠杆菌科细菌中最常见的自主复制接合型质粒,而且其在耐药基因快速水平散播过程中也发挥了至关重要的作用[18]。目前,人们对少数质粒接合作用的调控方式有了初步的认识和了解,但对于IncFⅡ质粒接合作用的调控方式尚不清楚,而且也不明白诸多调控蛋白之间的互作调控机制。
本研究利用Red同源重组的方法,将大肠杆菌ATCC25922的cpx R和hns基因敲除,用来研究调节蛋白H-NS及双组分信号转导系统Cpx AR 互作调控IncFⅡ质粒接合作用的分子机制,但是改变单一基因的表达量,往往达不到预期的理想效果。所以本研究又进一步在cpx R的基础上再次利用此方法缺失hns基因,构建双基因缺失株,以及它们的缺失回补菌株,在对单基因缺失△cpx R的PCR 检测时,用了CR-F/CR-R、KF/KR、CR-F/KR、KF/CRR 共4对引物。CR-F/CR-R 是1 对包含cpx R较长片段的引物,用其扩增缺失前菌株和kanr基因替代cp x R基因后的菌株都获得了预期大小的片段,缺失前为1 356 bp,同源重组后为2 407 bp,KF/KR是从kanr基因内部选取一段引物,用来检测同源重组后打靶片段成功替换cpx R基因。CR-F/KR、KF/CR-R 用于检测kanr基因插入的位置,用以上引物扩增都获得了预期大小的片段。用p CP20 质粒去除kanr片段,筛选出来的重组子用CR-F/CR-R扩增,得到与预期一致的目的条带,测序结果显示,只剩下很短的(83 bp)残留的FRT 位点,证明cpx R被成功敲除。本试验设计的4对引物能更明显更直观的检测出缺失株构建是否成功,以及同源重组的位置是否正确。
本研究利用Red 同源重组技术,成功构建cpx R和hns的单/双基因缺失株及相应的回补株,为构建IncFⅡ质粒的各种重组菌提供了有力的保障,并为进一步研究H-NS 及Cpx AR 互作调控IncFⅡ质粒接合作用的分子机制奠定基础。
