地聚合物凝胶体系中N-A-S-H和C-A-S-H结构的分子模拟
2020-03-12康升荣吴丽梅
王 晴, 康升荣, 吴丽梅, 唐 宁, 张 强
(沈阳建筑大学 材料科学与工程学院, 辽宁 沈阳 110168)
地聚合物被认为是可以取代传统水泥的绿色凝胶材料,其反应产物的结构以硅铝四面体形成的无定形结构为主,根据含钙量不同可分为高钙体系(C-A-S-H)和低钙体系(或无钙体系)(N-A-S-H)[1].
低钙体系的地聚合物凝胶主要为无定形态的N-A-S-H凝胶,其化学组成和分子结构类似于天然沸石[1].Behzad[2]研究认为地聚合物凝胶结构中Si和Al元素均以Ⅳ配位形式存在,其中[AlO4]5-四面体需依靠结构空隙中的阳离子(Na+、Ca2+等)来平衡结构整体的电负性.张云升等[3]研究表明,在地聚合物中Si主要以SiQ4(2Al)和SiQ4(4Al)的形式存在,Al以AlQ4(4Si)的形式存在.根据Rovnaník[4]的研究,生成N-A-S-H的反应式为SiO2·Al2O3+4OH-+3H2O→2[Al(OH)4]-+[SiO2(OH)2]2-.
高钙体系的地聚合物凝胶主要为无定形态的C-A-S-H凝胶,具有和水化碳酸钙(C-S-H(I))相似的Tobermorite-14Å结构[5],由2层钙氧八面体和1层硅氧四面体构成层状结构[6],碱金属阳离子和H2O填充层间位置.一般认为Al进入C-S-H结构内部取代部分Si形成无定形状态的C-A-S-H, Faucon 等[7]研究表明Al有2种方式进入C-S-H结构内部取代部分Si,形成无定形的C-S-H,即配位四面体和桥接四面体的Al取代Si.
地聚合物制备过程中原材料的化学组成及性质对产物的种类和结构影响比较复杂[8].为研究碱胶凝材料水化产物的微观结构与性能,一般以人工合成的方法制备C-A-S-H和N-A-S-H凝胶[9],通过X射线衍射(XRD)、魔角自旋核磁共振技术(MAS-NMR)、扫描电镜/能谱仪(SEM/EDX)、透射电镜/能谱仪(TEM/EDX)和高分辨电子显微镜(HREM)等研究合成凝胶的化学组成和微观结构,进而预测地聚合物的宏观性能[10].N-A-S-H的合成方法主要有溶胶-凝胶法、碱溶液激发前驱体粉末法[11]和水热法合成法;C-A-S-H的合成方法主要有水热合成法、溶液反应法、有机载体合成法和单矿水化反应法等[8].
分子模拟是最热门的一种计算机模拟材料的研究方法,其中分子动力学模拟是目前凝聚态纳米尺度物理分析中应用较为广泛的一种计算方法[12],以统计力学为基础,建立微观量和宏观量或者可测量之间的联系.地聚合物凝胶结构分子模型的构建一般有2种方法,一是通过对已知类沸石结构进行结构放松和原子取代构建模型,二是蒙特卡罗方法的随机建模.例如Fernndez-jiménez等[13]和施惠生等[14]以Na原子、H2O分子和 Si2AlO10基团作为基本单元,根据试验数据和N-A-S-H凝胶结构的特点,模拟构建了N-A-S-H凝胶模型.周崇松[15]、Puertas等[16]、Hou等[17]、马骁等[18]用Al代替Si建立C-A-S-H凝胶模型,并研究了其结构的机械性能.
本文利用Materials Studio软件,构建了N-A-S-H和C-A-S-H凝胶结构模型.在Universal力场下对凝胶结构模型进行了结构优化和分子动力学模拟,对比分析N-A-S-H和C-A-S-H凝胶体系的能量统计特征、温度变化、径向分布函数,模拟体系的弹性模量和XRD图谱,并与溶胶-凝胶法制备的N-A-S-H和C-A-S-H凝胶进行对比.
1 原材料与试验
1.1 原材料
硅酸铝(Al2O3·SiO2,分析纯)购自上海麦克林试剂公司;氧化钙(CaO,分析纯)、氢氧化钠(NaOH,分析纯)购自天津市恒兴化学试剂制造有限公司,水玻璃购自沈阳富保佳化工原料公司,固含量(1)文中涉及的固含量、硅铝比等除特别说明外均为质量分数或质量比.为29.8%,模数为3.0.
1.2 材料表征方法
傅里叶红外光谱分析(FTIR)采用美国 Thermo Nicole的Nexus智能傅里叶变换红外光谱仪,试验样品采用压片法,样品与溴化钾质量比为1∶100.测量范围为4000~400cm-1.
X射线衍射分析(XRD)采用日本SHIMADZU公司生产的XRD-7000型X-射线衍射仪,扫描速度为5(°)/min,用连续扫描的方式扫描5°~90°范围内的衍射峰.
1.3 地聚合物的制备方法
相关研究表明[19],当硅铝比接近2时,地聚合物具有较好的强度,所以本试验控制原材料中硅铝比为2.
N-A-S-H凝胶的制备.首先按照配合比将 16.2g 的Al2O3·SiO2与145.6g的水混合制得硅铝质水溶液,接着以水玻璃作为激发剂和硅源,在81.2g的水玻璃中加入9.1g NaOH调节激发剂模数至1.4,然后缓慢地将硅铝质溶液滴入配制好的水玻璃溶液中,边滴加边用玻璃棒搅拌,搅拌完成后静置48h,再用蒸馏水洗涤过滤3次,制得N-A-S-H凝胶,烘干备用,烘干温度控制在80~90℃.
C-A-S-H凝胶的制备.采用和N-A-S-H凝胶相同的制备方法,不同的是加入水的质量为193.6g,加入的CaO为12.0g,制得C-A-S-H凝胶,烘干备用,烘干温度控制在80~90℃.C-A-S-H凝胶中CaO摩尔分数为30%,CaO摩尔分数按照下式计算:
(1)
式中:x(CaO)为CaO的摩尔分数;n(CaO),n(Al2O3),n(SiO2),分别为CaO,Al2O3和SiO2的物质的量.
1.4 分子动力学模拟
分子动力学模拟首先采用Materials Studio 7.0软件中的Forcite模块对体系进行几何优化.算法为Smart,能量收敛精度为0.0042kJ/mol,优化步数为1000,力场为Universal.体系的结构优化分3步,分别为Steepest、Quasi-Newton、Newton-Raphson,优化结束后体系能量值最低且收敛,表明结构已稳定.动力学计算采用Dynamics,范德华与静电作用能的求和方法分别为Atom based 与Ewald,截断半径设置为0.85nm.为得到体系能量最低构象,对几何优化后的结构进行等温等压(NPT)和正则(NVT)系宗下的退火动力学模拟.总模拟时长为100ps,退火温度为300K,退火循环次数为5次.分子动力学过程嵌入的数值求解方法为Verlet积分算法,用于求解牛顿运动方程.相关参数设置如 表1、 2所示.

表1 Geometric optimization结构优化参数设置
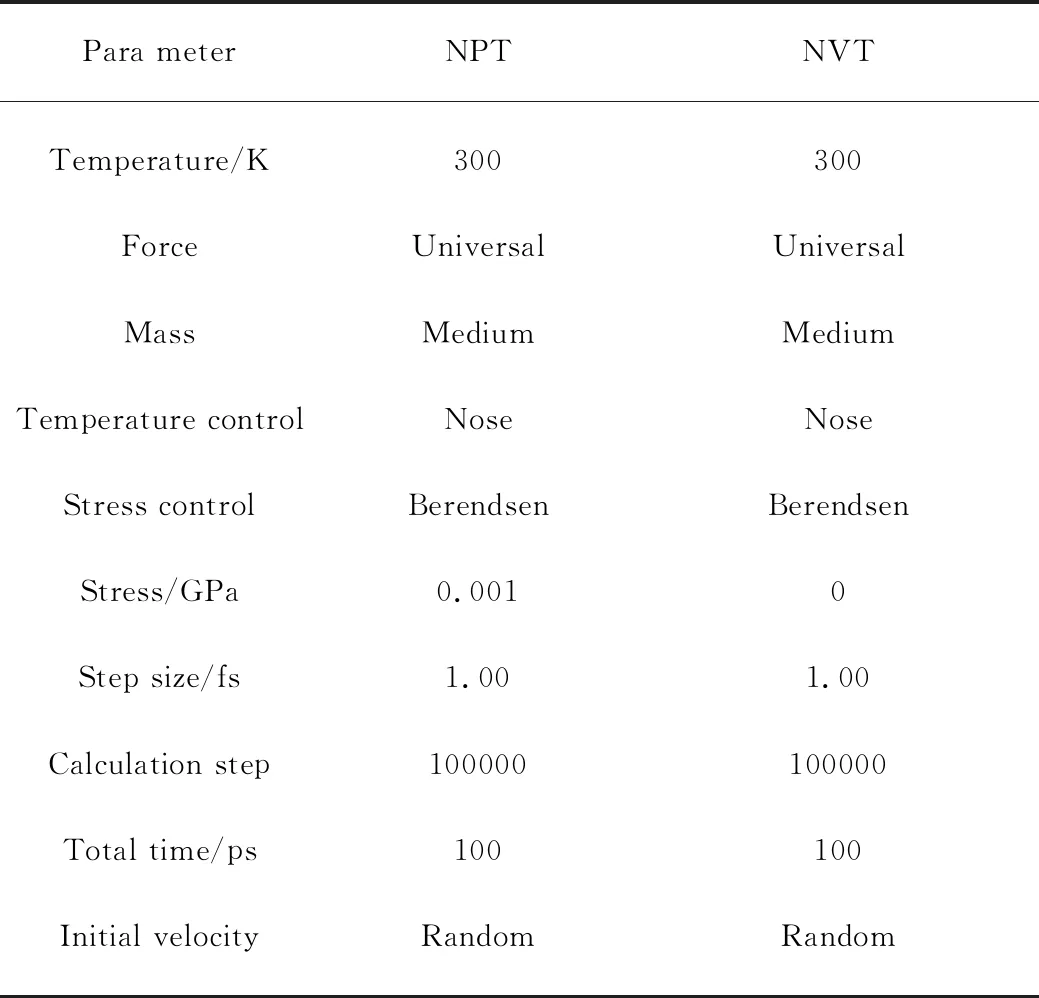
表2 分子动力学模拟参数设置
2 结果与讨论
2.1 凝胶结构模型的建立
地聚合物凝胶的三维周期性结构,运用Amorphous Cell模块建立,并用此模块的计算功能完成模拟单元的填充,构建Na原子、Ca原子、H2O分子、OH基团及Si2AlO10(Si—Al—Si)基团结构模型,并在DMol3模块中对所建H2O分子、—OH基团及 Si2AlO10(Si—Al—Si)基团结构模型进行几何优化.
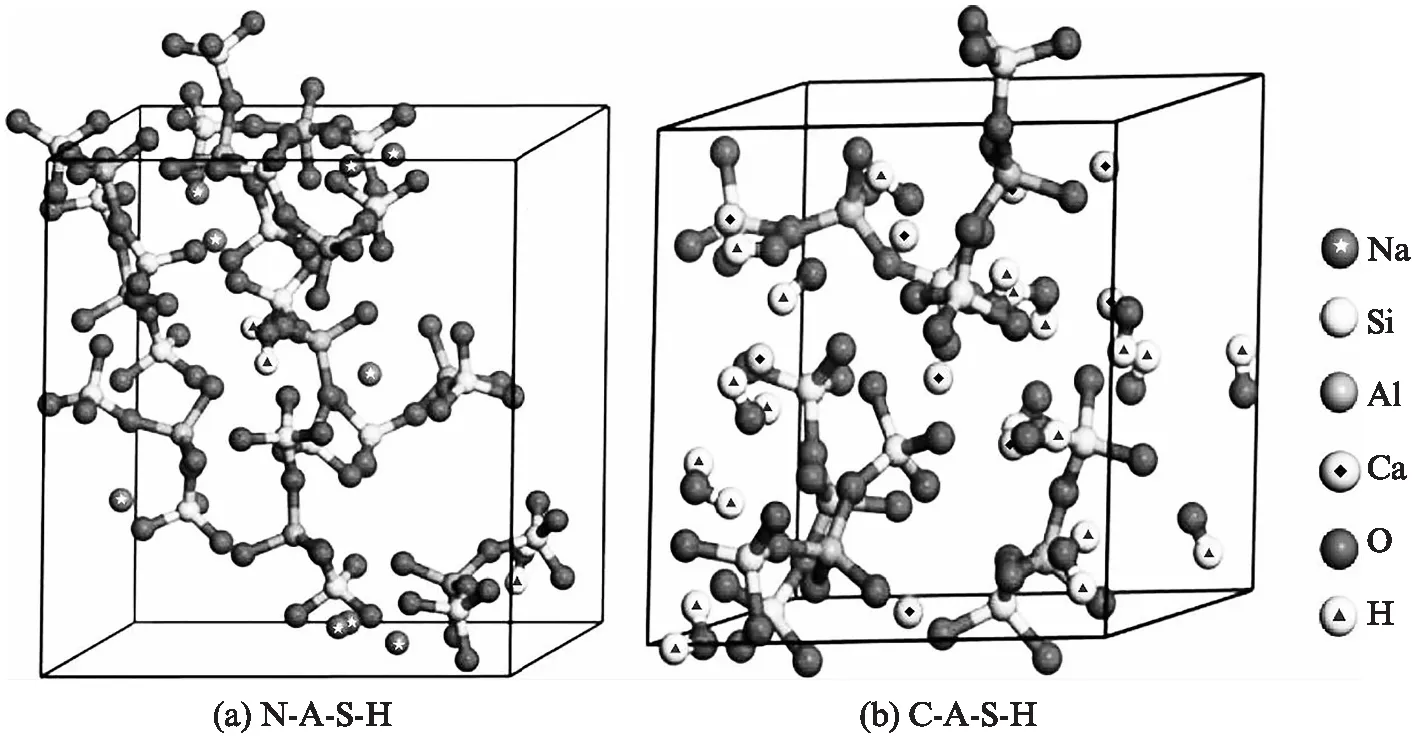
图1 地聚合物凝胶结构模型Fig.1 Cementitious structure simulation of geopolymer
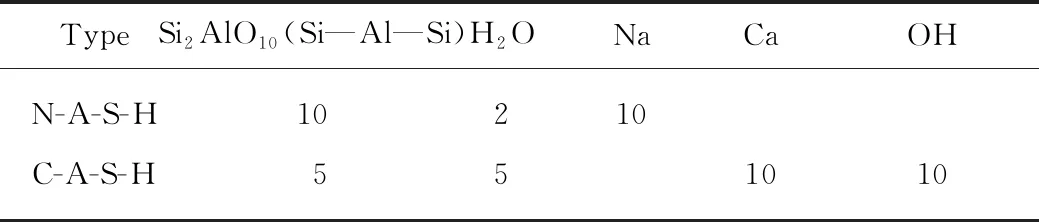
表3 地聚合物凝胶结构模型基本单元数
2.2 凝胶结构的稳定性
在分子动力学模拟过程中,根据观测能量和温度的变化可以判断结构的合理性和稳定性,一般认为当体系能量和温度的变化曲线波动在10%以内时,体系达到平衡条件.N-A-S-H和C-A-S-H凝胶体系的能量(E)变化曲线如图2所示.由图2可知,N-A-S-H和C-A-S-H凝胶体系的能量变化可以在100ps内趋于稳定,2种体系能量和温度在分子动力学模拟过程中均能够迅速达到平衡,说明模拟时参数设置较合理.
C-A-S-H凝胶体系的最终势函数能(potential energy)和非键作用能(non-bond energy)远低于初始值,而N-A-S-H凝胶体系的最终势函数能和非键作用能在初始值附近波动且低于C-A-S-H凝胶体系.经NVT系综分子动力学模拟后,N-A-S-H和C-A-S-H凝胶结构模型最终密度分别为1.075、 1.684g/cm3.
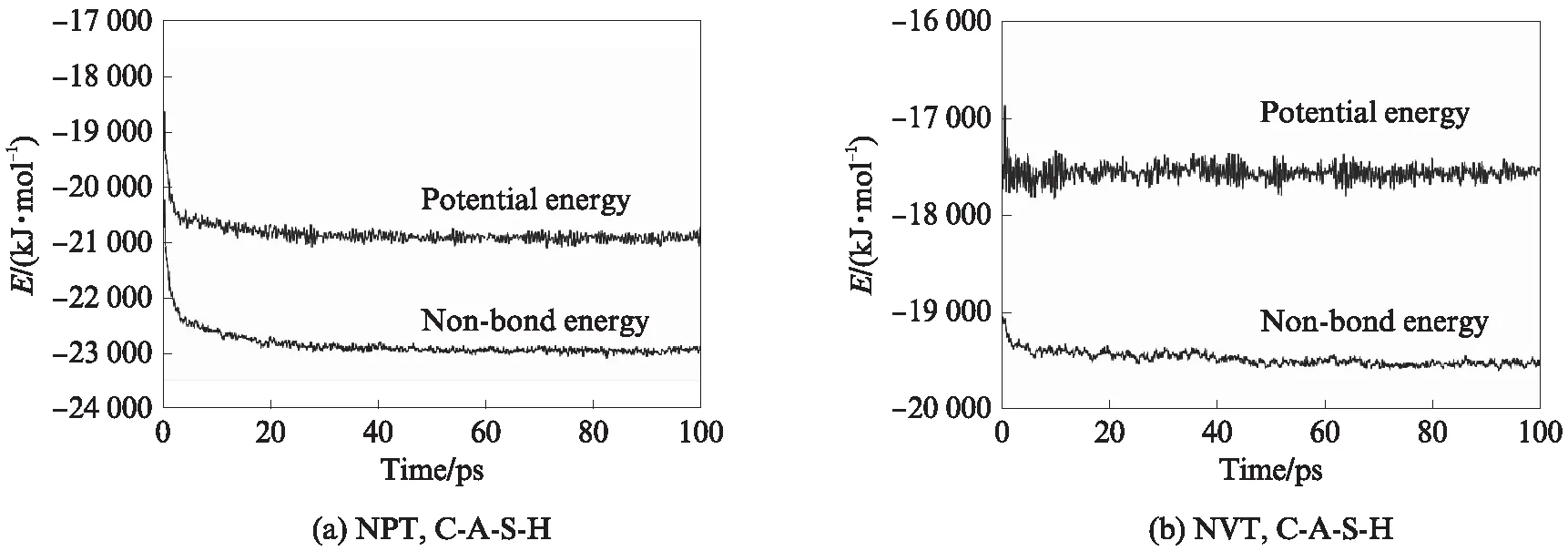
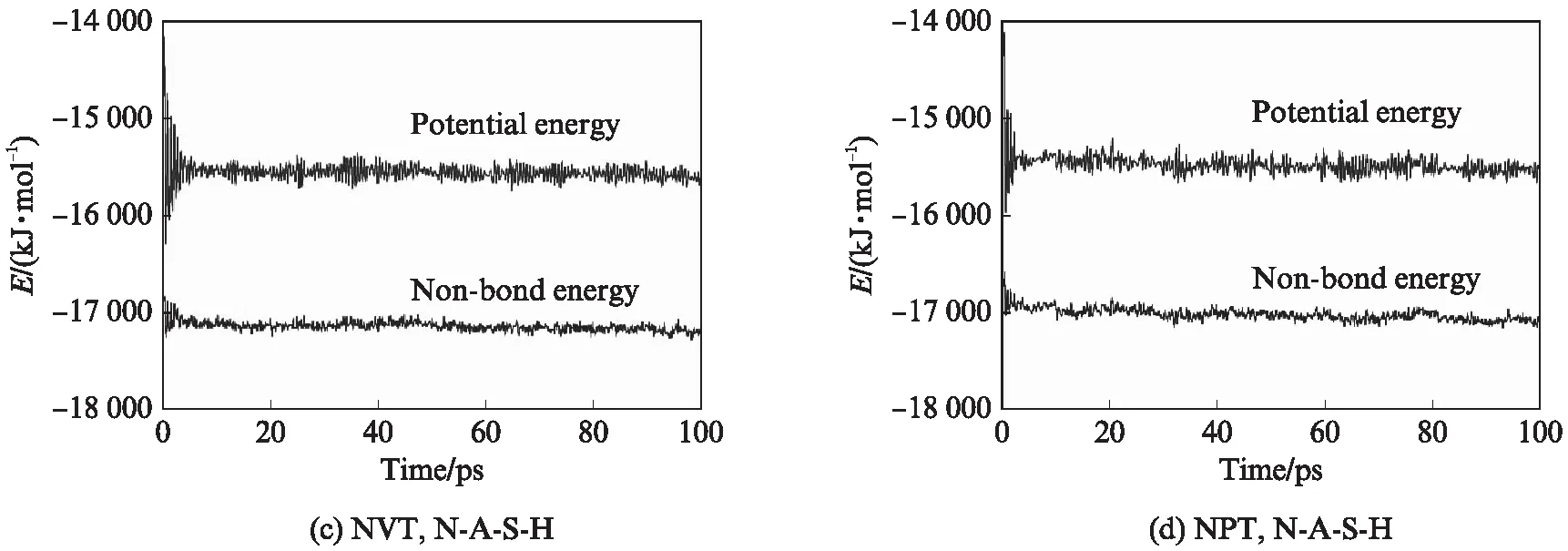
图2 N-A-S-H和C-A-S-H凝胶结构模型的能量变化Fig.2 Energy change of N-A-S-H and C-A-S-H with different simulations
N-A-S-H和C-A-S-H凝胶体系的温度变化曲线如图3所示.由图3可知,N-A-S-H和C-A-S-H凝胶体系的初始设定温度为300K,经过100ps后,体系温度一直在300K上下小范围内浮动并趋向于稳定.
2.3 凝胶结构的微观表征
N-A-S-H和C-A-S-H凝胶的FTIR图谱如图4所示,相关特征吸收峰统计见表4.N-A-S-H和C-A-S-H凝胶的主要吸收峰位置在476、740、 1091cm-1附近,其中476cm-1处为O—Si—O非对称伸缩振动,740cm-1处为O—Al—O非对称伸缩振动, 1091cm-1为Si—O—Si的非对称伸缩振动.N-A-S-H凝胶在615、874cm-1处有Si—O—Si面内弯曲振动,C-A-S-H凝胶在880cm-1处有Si—O—Si面内弯曲振动,说明地聚合物凝胶主要结构由 Si— O—Si和O—Al—O官能团组成.N-A-S-H和C-A-S-H凝胶在图中1630、3756cm-1处的吸收峰,分别为吸附H2O弯曲振动和游离H2O的伸缩振动,说明所制备的凝胶结构中不但含有游离 H2O,而且还含有吸附H2O,且N-A-S-H凝胶中O1和P1处吸收峰较C-A-S-H凝胶中O2和P2处吸收峰强.此外,对比N-A-S-H凝胶,在C-A-S-H凝胶FTIR图谱 中1384cm-1(Q)处出现了结合水 (—OH) 面内弯曲振动,说明C-A-S-H凝胶中含有结合水(—OH).因此,建模时C-A-S-H凝胶基本结构单元包含—OH基团,而N-A-S-H凝胶基本结构单元不含—OH基团的选择是符合实际的.
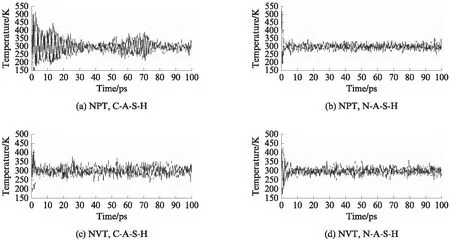
图3 N-A-S-H和C-A-S-H凝胶结构模型的温度变化Fig.3 Temperature change of N-A-S-H and C-A-S-H with different simulations
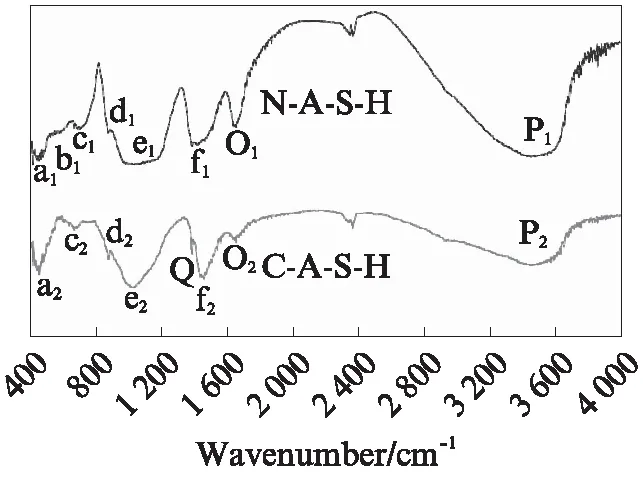
图4 N-A-S-H和C-A-S-H凝胶结构的红外吸收图谱Fig.4 FTIR spectra of N-A-S-H and C-A-S-H
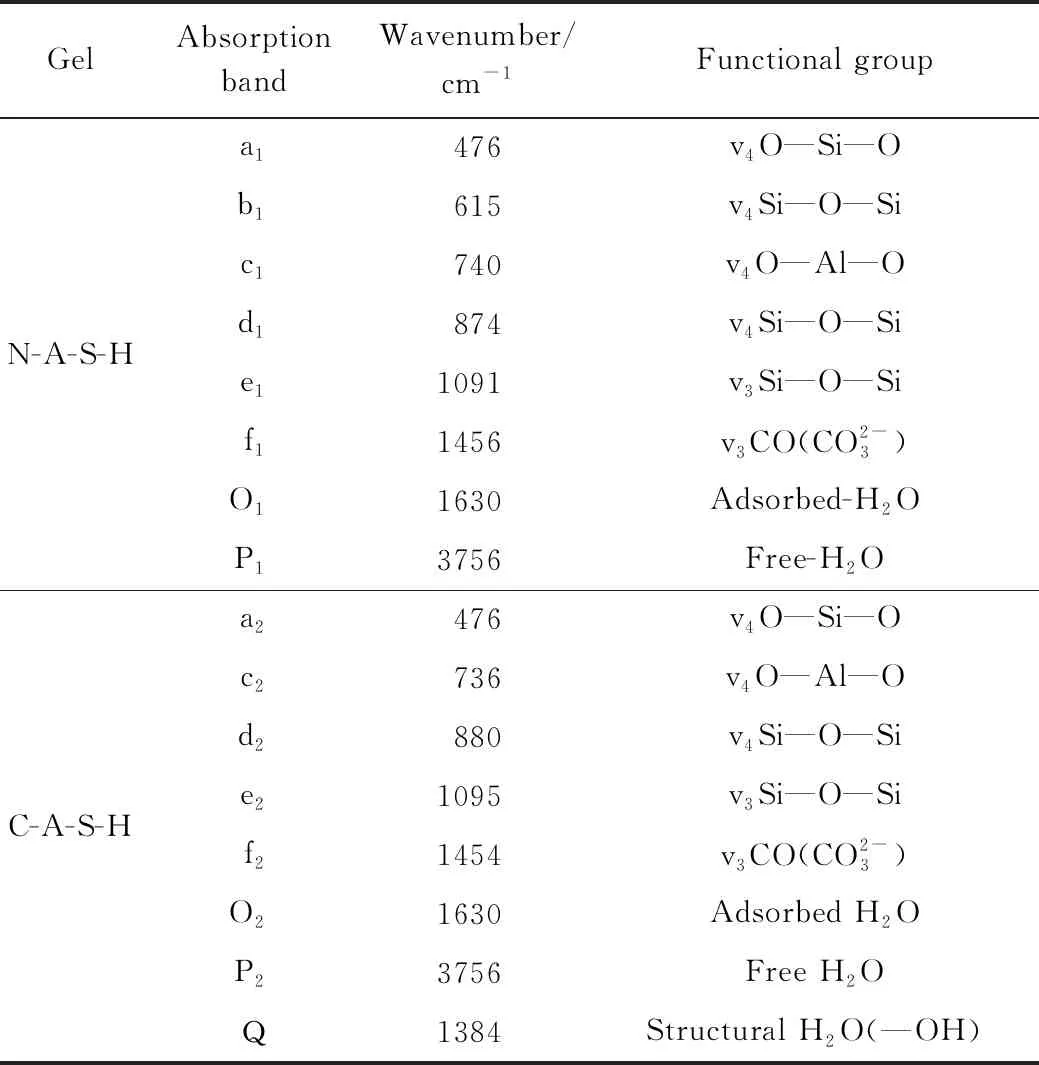
表4 N-A-S-H和C-A-S-H凝胶结构的红外光谱特征峰
N-A-S-H和C-A-S-H凝胶的XRD图谱如图5所示.由图5可见,试验制备的N-A-S-H凝胶的衍射峰为2θ=15°~35°范围的弥散峰,峰值在26°左右;C-A-S-H凝胶的衍射峰为2θ=20°~40°范围的弥散峰,峰值在30°左右,说明所制备的地聚合物凝胶主要为无定形态.N-A-S-H凝胶烘干后主要检测到的物质为Si、Al、Na的氧化物与H2O分子构成的未知物质(图中N-A-S-H试验的1、2、3)和1.2nm贝德石(图中N-A-S-H试验的4);C-A-S-H凝胶主要检测到方解石(图中C-A-S-H试验的1)、钙硅石(图中C-A-S-H试验的2)、氢氧钙石(图中C-A-S-H试验的3)和C-S-H(图中C-A-S-H试验的4).方解石为原料中CaO与水反应生成Ca(OH)2,Ca(OH)2再与空气中的CO2所形成,钙硅石来源于反应早期水玻璃和CaO的作用.此外,C-A-S-H凝胶中检测到C-S-H的存在,而N-A-S-H凝胶中没有C-S-H,这与FTIR测试时只有C-A-S-H凝胶中含有结构水(—OH)的结论一致.表明地聚合物中无定形态体系出现了向晶体转变的特征.
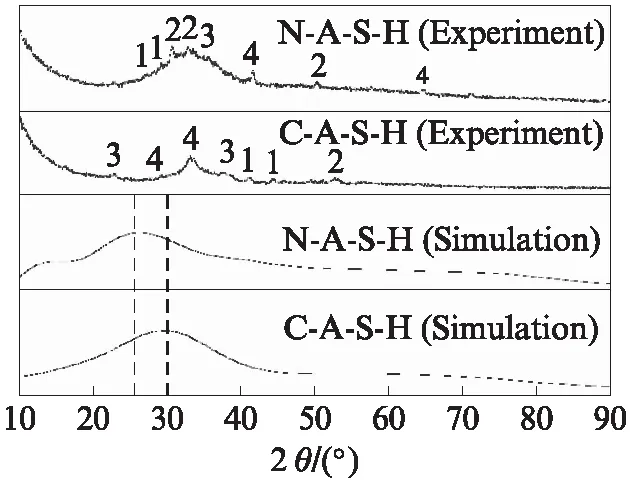
图5 N-A-S-H和C-A-S-H凝胶结构的XRD图谱对比Fig.5 Experimental and simulated results comparison of N-A-S-H and C-A-S-H based on XRD patterns
图5中,N-A-S-H凝胶结构模型的衍射峰为 2θ= 11°~33°范围的弥散峰,峰值在21°左右.根据Rovnaník[4]的研究,85℃以上结构无序的N-A-S-H会转化为结晶态的沸石相结构,所以XRD图谱弥散峰峰值位置试验值相较于模拟值偏大5°左右.C-A-S-H凝胶结构模型的衍射峰为2θ=20°~40°范围的弥散峰,峰值在30°左右,与试验制备的C-A-S-H凝胶的XRD衍射峰宽度和峰值位置一致.
2.4 凝胶结构的动力学轨迹
对凝胶结构模型进行分子动力学模拟,可以获得体系运动轨迹,进而可以计算体系的径向分布函数g(r).本文计算了N-A-S-H和C-A-S-H凝胶结构模型分子动力学模拟后的总径向分布函数和体系中Si—O、Al—O、Ca—O和O—O键在0~1.0nm范围的径向分布函数,结果如图6所示.由图6可见,N-A-S-H和C-A-S-H凝胶结构模型总径向分布函数峰在0~0.5nm之间,0.5~1.0nm范围内g(r) 趋近于1,说明原子间排布近程有序,远程无序,结构体系模型为典型的无定型结构,符合地聚合物凝胶结构的特点.N-A-S-H凝胶模型中Si—O、Al—O和O—O键径向分布函数第一峰位置附近峰型未分裂,而C-A-S-H凝胶中Si—O、Al—O、Ca—O和O—O键的第一峰位置附近出现了明显的分裂现象,说明C-A-S-H中Si—O、Al—O、Ca—O和O—O键成键类型较N-A-S-H中更加多样化,C-A-S-H体系更趋于致密;且C-A-S-H中Ca—O键径向分布函数出现了多个Ca—O峰,峰型均比其他原子对的峰更宽更低,说明Ca可以和距离本身较远的O形成Ca—O键,Ca—O键的成键范围比其他原子对成键范围更广.
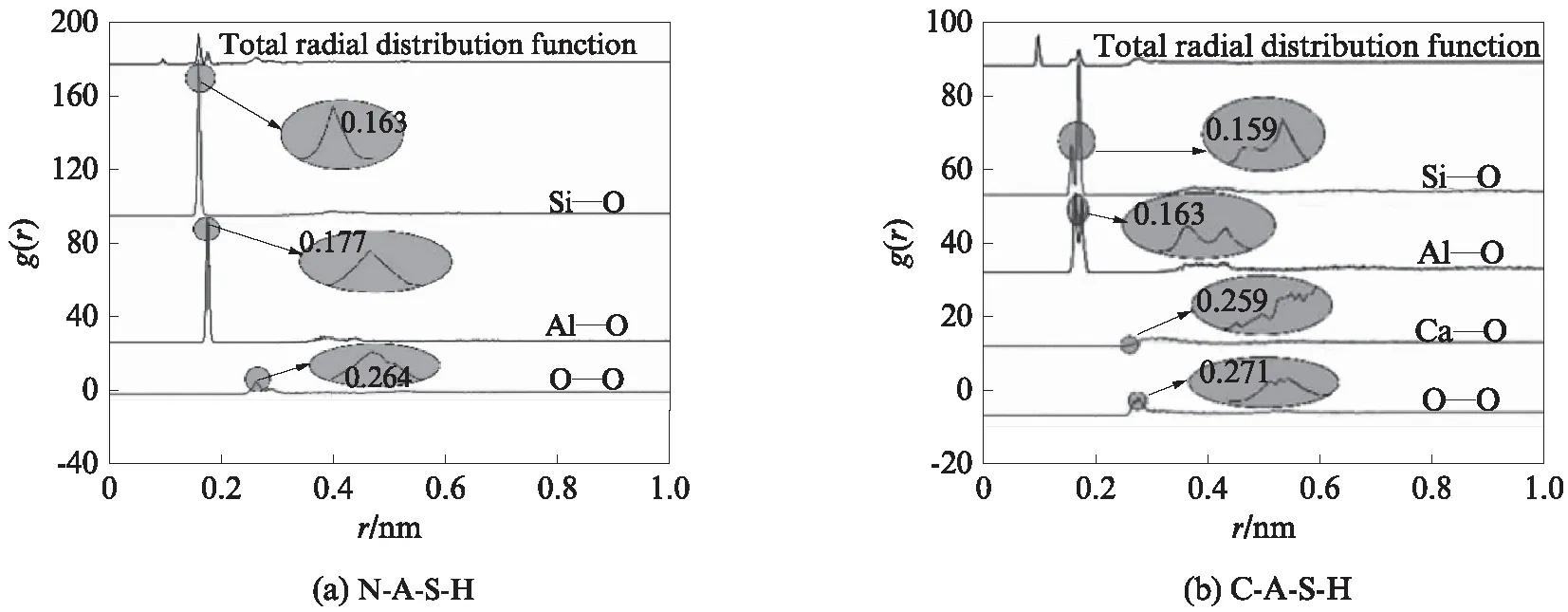
图6 N-A-S-H和C-A-S-H凝胶结构模型的径向分布函数Fig.6 Radial distribution of N-A-S-H and C-A-S-H after simulation
根据各原子对径向分布函数第一峰所处位置可以求得其平均键长,N-A-S-H凝胶结构模型在Universal力场下各原子间平均键长统计结果见表5.对比施惠生等[14]在Compass力场下构建的结构模型和White等[21]通过试验测得的N-A-S-H凝胶各原子间键长可知,所建模型除H—O键平均键长稍大于试验值外,Si—O和Al—O键均符合实际键长.由此可见,在Universal力场下构建并通过结构优化和分子动力学模拟所获得的N-A-S-H凝胶结构模型符合地聚合物中N-A-S-H凝胶的结构要求,验证了所建结构模型的准确性,可以利用该结构来进行N-A-S-H凝胶的研究.C-A-S-H凝胶结构模型原子间平均键长见表5,对比周崇松[15]对Al取代C-S-H中Si构建的C-A-S-H凝胶结构模拟结果及Smith等[22]和Merlino等[23]通过试验测得的C-S-H凝胶各原子间键长可知,所建模型Al—O、O—O、Si—O和Ca—O键的平均键长接近试验值,结果表明在Universal力场下构建并经结构优化和分子动力学模拟所获得的结构模型符合C-A-S-H凝胶的结构要求.
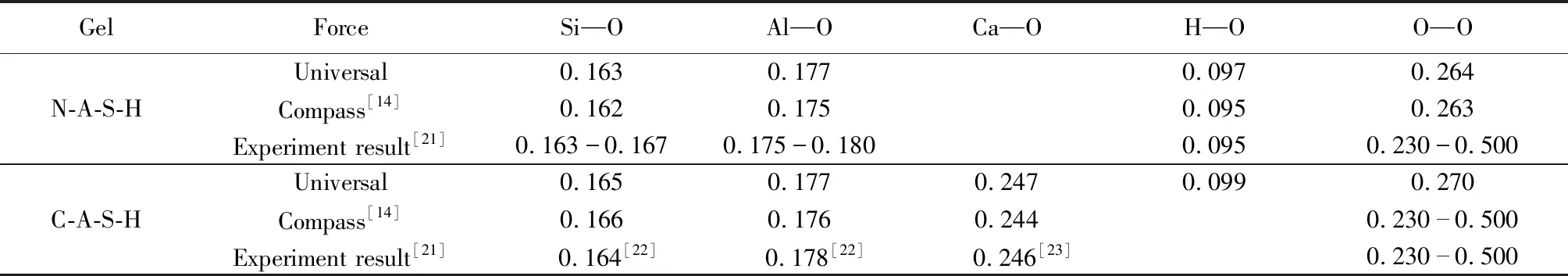
表5 N-A-S-H和C-A-S-H凝胶结构模型在Universal力场下原子间平均键长
2.5 凝胶结构的弹性模量
分子动力学模拟的(以NVT系综下结果为最终值)N-A-S-H和C-A-S-H凝胶结构模型在Universal力场下的模拟弹性模量见表6.由表6可见,N-A-S-H凝胶结构模型弹性模量的Universal力场模拟值较Compass力场模拟值偏大,但2种方法模拟结果均符合实际测量结果.C-A-S-H凝胶结构模型弹性模量为62.37GPa,比由Tobermorite结构掺杂Al后在Compass力场下的弹性模量模拟值更接近于Pellenq[25]实际测量结果,且对比N-A-S-H弹性模量模拟值可知C-A-S-H凝胶弹性模量远高于N-A-S-H凝胶.

表6 N-A-S-H和C-A-S-H凝胶结构模型的弹性模量
3 结论
(1)N-A-S-H和C-A-S-H凝胶结构模型的能量温度变化曲线能够迅速达到平衡,模拟结构稳定性好;且动力学轨迹、微观表征及弹性模量的模拟值与试验值或文献值吻合较好,证明了所建模型的有效性.
(2)由于钙元素含量的增加,C-A-S-H凝胶体系的能量增加到了1.684g/cm3,比N-A-S-H凝胶体系的能量1.075g/cm3提高了56.7%;地聚合物的无定形凝胶体系出现了向晶体转变的特征.
(3)动力学轨迹表明Ca—O键的成键范围比其他原子对成键范围更广,且C-A-S-H凝胶体系更趋近于致密;C-A-S-H凝胶体系的弹性模量明显高于N-A-S-H凝胶体系,对地聚合物的力学性能产生正向增益.
