一个伴不完全外显机制的Alagille综合征家系的遗传学诊断
2020-03-08胡亚倩郑必霞刘志峰李玫金玉张志华
胡亚倩,郑必霞,刘志峰,李玫,金玉,张志华
(南京医科大学附属儿童医院 消化科,江苏 南京 210000)
Alagille综合征(ALGS)是一种常染色体显性遗传病,主要发生于儿童和青少年,病因为JAG1或Notch2 基因突变影响Notch信号通路,从而引起肝内小叶间胆管减少[1]。临床上多因婴儿期胆汁淤积症就诊,常伴有心脏疾病、骨骼异常、眼部异常和特殊面容等临床表现。作者回顾一个家系中两例ALGS患儿的临床表现及基因检测结果,并复习相关文献,总结ALGS的临床特征及遗传特点,为临床更好地选择分子诊断方法并早期识别、诊治ALGS提供思路。
1 病例介绍
1.1 病例一般情况
患儿之兄,6岁,5年前因“发现肝功能异常伴皮肤瘙痒1月余”至外院就诊。大便黄色。查体:眼眶凹陷、前额突出、鼻梁低平、尖下颌,皮肤、巩膜无明显黄染(图1左);心前区可及3/6级收缩期杂音,无传导;肝脏肋下2.5 cm、剑突下2.0 cm,质软,边缘光整,无压痛;脾脏肋下未触及。间断口服“保肝、利胆、退黄”药物治疗。目前身高113.0 cm,体重17 kg,头围48.5 cm。
患儿,2岁7个月,因“反复皮肤瘙痒1年余”入院。大便黄色。查体:头围45.8 cm,身高85 cm,体重10 kg,眼距稍宽,下颌尖小,皮肤及巩膜未见黄染,(图1右);心前区全收缩期3/6级杂音,无传导;肝脏肋下2.0 cm、剑突下3 cm,质软,表面光滑;脾脏肋下未触及。

图1 患儿之兄(左)及患儿(右)
父母否认类似病史,否认家族遗传疾病史。
1.2 临床检查
1.2.1 检查方法
患儿兄既往在外院行JAG1基因一代测序检测。
采集患儿及其兄、父、母静脉血3 ml(EDTA抗凝)送至北京全谱医学检验实验室,进行全外显子组测序,外周血标本DNA抽提前,患儿监护人填写知情同意书,本研究获南京儿童医院医学伦理委员会批准。使用血液基因组柱式中量提取试剂盒(康为世纪生物科技有限公司)提取基因组DNA,操作按照试剂盒说明书进行。采用IDT公司xGen®Exome Research Panel v1.0捕获探针与gDNA文库序列进行液体杂交,将目标区域DNA片段进行富集,构建全外显子文库,通过Illumina公司 NovaSeq 6000系列测序仪进行高通量测序(PE150),目标序列测序覆盖度不低于99%。测序过程由智因东方转化医学研究中心完成。
对检测出的致病可能性较高的基因变异,采用荧光定量PCR(qPCR)的方法进行验证。qPCR的引物序列设计见表1。

表1 qPCR扩增的引物序列
对检测出的大片段拷贝数变异,进一步采用CNV-seq分析。经DNA提取、打断、文库构建步骤,并经Illumina Hiseq系列测序仪测序完成,由Illumina官方basecall分析软件BclToFastq得到原始数据,经检测、过滤后由全谱®遗传病精准诊断云平台系统分析筛选,结合Decipher、ClinVar、OMIM、DGV、ClinGen、ISCA等数据库比对,并结合剂量敏感性及临床特征吻合度等要素综合分析,对CNV的致病性进行分级。
1.2.2 临床实验室检查结果
1.2.2.1 患儿之兄 2014年5月15日:血生化:丙氨酸氨基转移酶1 353.0 U·L-1,天门冬氨酸氨基转移酶953.0 U·L-1,谷氨酰转肽酶537.0 U·L-1,肌酸酶同功酶53.2 U·L-1,碱性磷酸酶 736.6 U·L-1,总胆红素36.0 μmol·L-1。经保肝、利胆退黄治疗后2014年5月21日复查:血生化:丙氨酸氨基转移酶 435.0 U·L-1, 天门冬氨酸氨基转移酶 234.0 U·L-1,谷氨酰转肽酶 295.0 U·L-1,肌酸酶同功酶41.0 U·L-1, 碱性磷酸酶544.0 U·L-1,总胆红素35.71 μmol·L-1,直接胆红素29.19 μmol·L-1,总胆固醇8.18 mmol·L-1,胆汁酸329.0 μmol·L-1;巨细胞病毒(CMV)、EB病毒(EBV)DNA阴性;EB病毒抗体IgM阴性;TORCH:风疹病毒抗体IgG阳性,余阴性;凝血功能正常;传染病4项阴性、铜蓝蛋白正常;X线胸片:椎骨未及明显骨质异常;心脏彩超:左右肺动脉狭窄、卵圆孔未闭;肝胆胰脾B超未见明显异常。2017年10月17日门诊随访:血生化:丙氨酸氨基转移酶267.0 U·L-1,天门冬氨酸氨基转移酶164.0 U·L-1,谷氨酰转肽酶 456.0 U·L-1,总胆红素26.87 μmol·L-1,直接胆红素18.19 μmol·L-1,总胆固醇8.91 mmol·L-1。
1.2.2.2 患儿 2019年9月17日:血生化:丙氨酸氨基转移酶115.0 U·L-1,天门冬氨酸氨基转移酶102.0 U·L-1,碱性磷酸酶576 U·L-1,谷氨酰转肽酶316 U·L-1,总胆红素19.0 μmol·L-1,直接胆红素12.7 μmol·L-1,总胆固醇7.31 mmol·L-1,总胆汁酸 41.6 μmol·L-1;传染病4项、凝血功能、体液免疫、细胞免疫基本正常;EB-DNA、CMV-DNA阴性;EBV抗体、CMV抗体IgM阴性,铜蓝蛋白正常;心脏彩超:(1) 先天性心脏病:房间隔缺损;(2) 主动脉瓣轻度狭窄;(3)永存左上腔静脉;肝胆胰脾B超:肝脏形态欠规则,右叶斜径82 mm、右肋下21 mm;脾脏肋下可及,肋间厚22 mm、肋下19 mm,脾内光点均匀分布;心电图:窦性心律,P-R间期延长,右心负荷重?Q-T间期延长;X线胸片:心影饱满,两肺未及明显异常,椎骨未及明显异常。
1.2.3 基因检测结果
1.2.3.1 患儿之兄 JAG1基因检测(外院、Sanger测序)阴性。
1.2.3.2 患儿 采用芯片捕获高通量测序的方法对先证者全外显子组进行测序,结果提示:患儿chr20:10018949-10654178区段疑似存在约635 K大小的杂合缺失,缺失片段中包含完整的JAG1基因,进一步采用qPCR的方法验证其JAG1基因外显子杂合缺失。
1.2.3.3 家系中其他人 将正常对照样本与先证者及其兄、父、母样本进行同组qPCR检测,以ALB基因为内参基因,对目标基因JAG1的1,21和26号外显子的拷贝数进行检测。结果发现:先证者、先证者之兄及先证者之父JAG1基因1,21和26号外显子的拷贝数与正常对照的比值约为0.5,先证者之母JAG1基因1,21和26号外显子的拷贝数与正常对照的比值约为1.0(图2),提示先证者及其兄、父JAG1基因1-26外显子存在杂合缺失,先证者之母JAG1基因1-26外显子拷贝数正常。qPCR检测结果与全外显子组测序结果一致,提示患儿及其兄都存在JAG1基因外显子杂合缺失,且两兄弟基因杂合缺失来源于其父亲。
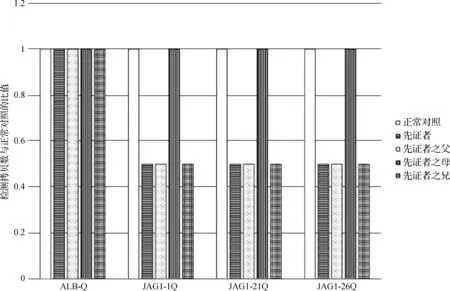
图2 JAG1基因外显子1-26杂合缺失突变qPCR验证结果 ALB-Q:内参基因;JAG1-1Q:JAG1基因外显子1;JAG1-21Q:JAG1基因外显子21;JAG1-26Q:JAG1基因外显子26
进一步采用CNV-seq检测对患儿及其父亲携带的累及JAG1基因的杂合缺失进行精确定位,发现患儿及其父亲chr20:9965785-11725865位置存在约1.76 Mb的可能致病性CNV,其中包含JAG1基因与Alagille综合征1型相关,按照ACMG变异评级标准,其生物及临床意义为致病性变异等级。
2 讨 论
ALGS(OMIM:118450)是一种罕见的常染色体显性遗传病,又称先天性肝内胆管发育不良综合征、动脉-肝发育不良,可累及肝脏、心脏、眼、骨骼、面部等多个系统,外显率94%左右,临床表现从没有症状到危及生命的情况均可发生[2]。
1997年Oda等[3]首次报道了JAG1基因突变引起ALGS。JAG1基因定位于染色体20p12,编码细胞膜表面蛋白Jagged1蛋白作为配体参与Notch信号通路[4-5]。2006年McDaniell等[6]报道了Notch2基因突变也可导致ALGS。Notch2定位于染色体1p11-p13,编码Notch2跨膜蛋白作为Notch信号通路的重要受体。NOTCH信号对胆管的起源至关重要,在ALGS的病理生理中起着重要作用[7-8]。研究表明94%~96%的ALGS由JAG1基因突变导致,Notch2基因突变占1%~2%,研究并未发现特定的突变和表达的表型之间有相关性[2]。
ALGS常包括心脏、肝脏、骨骼、眼部异常及特殊面容五大临床特征;也有学者把肾脏异常及血管畸形纳入主要临床特征。其他特点如身材矮小、发育落后、反复感染等亦可见。经典的ALGS诊断标准为肝组织活检有肝内小叶间胆管数量减少或缺如,具有至少5个主要临床表现中的3个,并排除其他可能原因。修订的ALGS诊断标准认为符合4个或以上主要临床表现也可诊断,如果已知有JAG1基因突变或阳性家族史时,1个以上主要临床表现通常即可确诊[9-10]。因此基因检测技术在该疾病诊断中的价值进一步凸显。本资料家系中患儿及其哥哥因皮肤瘙痒就诊,查体有肝脏增大、心脏杂音、特殊面容,辅助检查血生化显示肝功能异常、直接胆红素增高、γ-谷氨酰转肽酶增高、胆汁酸增高等胆汁淤积表现,心脏彩超可见肺动脉狭窄、主动脉瓣狭窄、房间隔缺损、室间隔缺损等异常。两兄弟同时存在肝脏胆汁淤积、心脏改变及特殊面容3项主要临床表现,但基因检测明确JAG1基因突变帮助尽早明确了ALGS的诊断。
治疗方面,ALGS目前无明确的病因治疗手段,主要为保肝降酶、利胆退黄、补充脂溶性维生素、维持营养等,定期监测脏器功能。一项对92例ALGS患者的研究[11]表明,随着年龄的增长,胆管狭窄和纤维化的发生率显著增加,最终导致肝硬化或肝衰竭而需要肝移植的患者大约为15%。Hadchouel[12]研究发现,肝移植后总的20年平均生存率为70%,Lee等[13]报道9例该病患儿肝移植后5及20年的生存率分别为88.9%和77.8%。严重胆汁淤积伴皮肤瘙痒的患儿经药物治疗无效后可考虑外科手术干预,阻断肠肝循环,但ALGS患者的胆管发育不全可导致到达肠道的胆汁减少,因此ALGS外科手术治疗效果通常比其他引起胆汁淤积的疾病治疗效果差;进行性胆汁淤积伴顽固性皮肤瘙痒的患儿可考虑肝移植[14-15]。Cardona等[16]报道,83.3%的ALGS患儿在活体肝移植后瘙痒消失,肝功能检查结果正常,提示严重肝型ALGS患儿肝移植后生活质量可有明显改善。本资料家系中患儿及其哥哥在给予阿拓莫兰、复方甘草酸苷保肝、熊去氧胆酸利胆退黄、补充脂溶性维生素等治疗后肝酶明显下降,但皮肤瘙痒改善不明显。长期随访如皮肤瘙痒加剧必要时可行手术治疗改善患儿生存质量。
患儿之兄5年前于外院就诊,一代测序检测JAG1基因阴性,但此次qPCR对其JAG1基因外显子验证发现患儿之兄存在JAG1基因的杂合缺失,提示一代测序的检测方法对基因组拷贝数变异的检测有一定的技术局限性。全外显子检测联合Sanger测序对确定点突变的位点更有价值;全外显子检测联合qPCR可用来检测出较小的变异并进行验证,但对大片段拷贝数变异的检测不够精确,CNV-seq检测可更精确地检测大片段拷贝数变异。因此,根据疾病的遗传特点及基因突变的情况选择合理的基因检测手段,既能帮助临床尽早明确病因和诊断、指导合理治疗并判断预后,也可为病人家庭减轻经济负担。
qPCR验证提示患儿及其兄JAG1基因杂合缺失来源于其父亲,但患儿父亲临床上无典型临床表现,符合JAG1基因外显率94%左右、可不完全外显的遗传特点[17]。该病不完全外显的特点为疾病的诊治带来了困难。与该疾病相关的肝脏、心脏、肾脏、眼部病变可单独或合并存在于其他综合征或疾病中[18],而某些满足ALGS临床特征的患者经基因检测却排除该疾病诊断,如Dyack等[19]报道的一个家系中5人患有Alagille-like综合征却为常染色体隐性遗传。Kamath等[20]首次报道了两例同卵双生子ALGS表型不一致的案例,提示非遗传因素在该疾病表型中有着重要作用。本资料家系中患儿父亲与患儿存在相同的拷贝数变异,但无典型的临床表现,也可能与非遗传因素影响疾病表型有关,因此临床对疑似ALGS的患者应适当放宽基因检测指征,以期尽早明确诊断。
CNV-seq检测发现,患儿及其父亲chr20:9965785-11725865位置存在约1.76 Mb的致病性CNV。该突变的发现丰富了JAG1基因变异谱。姜涛等[21]也报道了chr20p12.2缺失致ALGS患儿1例。WES等基因检测技术的发展和广泛应用使得更多基因突变类型得以检出,对疾病的认识得以进一步深入,反复胆汁淤积是儿科一个很棘手的问题,因此对反复胆汁淤积的患儿应尽早行基因检测明确诊断并早期提供遗传咨询与指导。
