基于第三代纳米孔测序技术的东方蜜蜂微孢子虫全长转录组构建及注释
2020-03-04陈华枝范小雪祝智威蒋海宾范元婵熊翠玲郑燕珍付中民徐国钧陈大福
陈华枝, 杜 宇, 范小雪, 祝智威, 蒋海宾, 王 杰, 范元婵,熊翠玲,2, 郑燕珍, 付中民,2, 徐国钧, 陈大福,*, 郭 睿,2,*
(1.福建农林大学动物科学学院(蜂学学院), 福州 350002; 2.福建农林大学蜂疗研究所,福州 350002)
东方蜜蜂微孢子虫Nosemaceranae是一种单细胞真菌病原,专性侵染成年蜜蜂的中肠上皮细胞,导致蜜蜂微孢子虫病(Traver and Fell, 2012)。该病原对西方蜜蜂Apismellifera幼虫同样具有侵染性(Eirietal., 2015)。东方蜜蜂微孢子虫可引起宿主出现寿命缩短、能量胁迫和免疫抑制等现象(Pariesetal., 2018)。此外,该病原还能与其他生物或非生物胁迫因子协同作用,严重影响蜜蜂健康、蜂群生产力和蜂产品产量(Wuetal., 2012; Shutleretal., 2014)。目前,世界各养蜂国家的蜂群均伴有不同程度的东方蜜蜂微孢子虫感染(Martín-Hernándezetal., 2018),且其毒力高低通常与季节变化密切相关(Gisderetal., 2010)。
Cornman等(2009)测序并公布了东方蜜蜂微孢子虫的基因组,为其分子生物学及组学研究奠定了基础。此后,人们利用第二代测序(second-generation sequencing)技术对西方蜜蜂响应东方蜜蜂微孢子虫胁迫的应答进行了一些转录组研究(Huangetal., 2015; Garridoetal., 2016; Badaouietal., 2017)。相对于西方蜜蜂,有关东方蜜蜂微孢子虫的组学研究较少(Huangetal., 2016; Huang and Evans, 2016)。来源于蜜蜂组织的可传代的体外培养细胞系缺失是一大限制因素。笔者所在团队前期已对东方蜜蜂微孢子虫进行了较为系统的转录组研究,在全基因组水平鉴定和分析了东方蜜蜂微孢子虫纯净孢子转录组中的长链非编码RNA(long non-coding RNA, lncRNA)(Guoetal., 2018b; 周丁丁等, 2020)和环状RNA(circular RNA, circRNA)(Guoetal., 2018a);通过全面分析东方蜜蜂微孢子虫侵染意大利蜜蜂Apismelliferaligustica工蜂过程的mRNA差异表达谱、miRNA差异表达谱及调控网络,揭示了东方蜜蜂微孢子虫的侵染机制(耿四海等, 2020a, 2020b);通过表达谱分析初步揭示了高表达基因在东方蜜蜂微孢子虫侵染中华蜜蜂Apisceranacerana工蜂中的潜在作用(熊翠玲等, 2020)。
牛津纳米孔(nanopore)技术是基于纳米孔的单分子实时电信号的第三代测序(third-generation sequencing)技术(Deameretal., 2016),其原理是DNA/RNA双链在马达蛋白的引导下与镶嵌在生物膜上的纳米孔蛋白结合并解螺旋,在生物膜两侧电压差的作用下,DNA/RNA链以一定的速率通过纳米孔通道蛋白,由于DNA/RNA链上不同碱基化学性质存在差异,故单个碱基或DNA分子通过纳米孔通道时会引起不同电学信号的变化(Magietal., 2018),通过对这些信号进行检测及对应,即可计算获得相应碱基的类型,完成序列的实时测定(Jainetal., 2016)。与基于边合成边测序原理的二代测序技术相比,以Oxford Nanopore和PacBio SMRT为代表的三代测序技术具有超长读长、较短的测序周期以及直接读取核酸修饰等优势(Luetal., 2016),因此近年来已成功应用于人类(Leaetal., 2018; Workmanetal., 2019)、穴兔Oryctolaguscuniculus(Chenetal., 2017)、果蝇Drosophila(Bayegaetal., 2020)、杨树Populus(Chaoetal., 2019)和辣椒Capsicumannuum(Zhuetal., 2018)等动植物的相关研究。但微生物的全长转录组研究相对缺乏且有限的研究多集中于病毒(Tombáczetal., 2017)。此前,笔者团队利用PaBio SMRT测序技术对纯培养蜜蜂球囊菌Ascosphaeraapis的纯化菌丝和纯化孢子进行测序,获得了高质量的全长转录本数据,并组装和注释了蜜蜂球囊菌的全长转录组(Chenetal., 2020)。这是迄今关于蜜蜂病原的唯一一例全长转录组研究报道。
本研究利用Oxford Nanopore测序技术对东方蜜蜂微孢子虫的纯净孢子进行转录组测序,基于高质量的测序数据组装东方蜜蜂微孢子虫的全长转录组,通过比对主流蛋白功能数据库对全长转录本进行功能注释,并通过生物信息学方法对lncRNA进行预测和分析,进而对全长转录本的表达量进行计算。研究结果可完善东方蜜蜂微孢子虫的转录组注释信息,为东方蜜蜂微孢子虫组学及分子生物学研究提供有价值的参考信息,也为基因全长克隆和功能研究奠定基础。
1 材料与方法
1.1 供试生物材料
本实验使用的东方蜜蜂微孢子虫侵染的意大利蜜蜂工蜂取自福州市闽侯县荆溪源安蜂场。前期研究中,笔者所在团队从实验蜂群中抓取外勤蜂,拉取中肠并纯化出病原孢子,显微镜视野下孢子呈椭圆形且折光性较强;同时,参照黄枳腱(2019)的方法对东方蜜蜂微孢子虫进行PCR鉴定。结合形态学观察和分子生物学验证结果证明该实验蜂群中意大利蜜蜂工蜂感染的真菌病原确为东方蜜蜂微孢子虫。
1.2 东方蜜蜂微孢子虫孢子样品的制备
按照笔者所在团队前期建立的方法(熊翠玲等, 2019a, 2019b)对东方蜜蜂微孢子虫孢子进行粗提和纯化:拉取感染工蜂的中肠,放入干净研钵,加适量无菌水研磨,经4层纱布滤除组织碎片;滤液经6 000 g离心5 min,用25%的Percoll溶液重悬沉淀;将100%, 75%, 50%和25%的Percoll溶液依次小心加入EP管,再将上述孢子粗提液轻缓加在Percoll不连续密度梯度溶液的液面之上,14 000 g离心30 min;用无菌的注射器针头小心吸取乳白色的孢子带,注入干净的EP管,液氮速冻后迅速转移至-80℃超低温冰箱保存备用。
1.3 cDNA文库构建及Nanopore全长转录组测序
利用TRizol试剂盒(Thermo Fisher公司,美国)提取1.2节东方蜜蜂微孢子虫孢子样品的总RNA;利用Maxima H Minus Reverse Transcriptase试剂盒(Thermo Fisher公司,美国)进行反转录,得到的cDNA加Switch Oligo,再合成互补链;DNA损伤修复和末端修复,磁珠纯化。委托北京百迈克生物科技有限公司对上述构建好的cDNA文库(命名为Nc)进行全长转录组测序,测序平台为Nanopore PromethION(Oxford Nanopore Technologies公司,英国)。
1.4 数据质控
Nanopore PromethION测序下机的raw reads格式为包含所有原始测序信号的二代FAST5格式;通过MinKNOW2.2软件包中的Guppy软件(Bayegaetal., 2020)对raw reads进行base calling,将数据转换为FASTQ格式;进一步过滤短片段和低质量的raw reads,得到高质量的clean reads。根据Nanopore cDNA测序原理(Jenjaroenpunetal., 2018; Boldogköietal., 2019),clean reads两端识别到引物则判定为全长转录本序列。
1.5 全长转录本的数据库注释
利用Blast工具将上述所有全长转录本比对Nr(non-redundant protein sequence database)(Dengetal., 2017), Swiss-Prot(The UniProt Consortium, 2017), KOG(eukaryotic ortholog groups)(Kooninetal., 2004), eggNOG(evolutionary genealogy of genes: Non-supervised Orthologous Groups)(Powelletal., 2014), Pfam(protein family)(Kanehisaetal., 2004), GO(Gene Ontology)(Ashburneretal., 2000)和KEGG(Kyoto Encyclopedia of Genes and Genomes)(McKennaetal., 2010)数据库比对,获得相应的注释信息。
1.6 lncRNA的鉴定
一般认为lncRNA不编码蛋白,因此可通过对转录本进行编码潜能筛选判定该转录本是否为lncRNA(熊翠玲等, 2018)。分别利用CPC(Kongetal., 2007), CNCI(Sunetal., 2013), CPAT(Wangetal., 2013)和Pfam(Finnetal., 2014)4种蛋白结构域分析方法对新发现的转录本进行lncRNA的预测。取四者的交集筛选高可信度lncRNA。
1.7 全长转录本的表达定量分析
全长转录组测序可以模拟成一个随机抽样的过程,为了让片段数目能真实地反映转录本表达水平,需要对样品中的mapped reads的数目进行归一化。采用CPM(counts per million)(Byrneetal., 2017)作为衡量全长转录本表达水平的指标,计算公式如下:
CPM=[(比对到转录本的reads数Reads mapped to transcript/样品中reads总数Total reads aligned in sample)×1 000 000]。
2 结果
2.1 全长转录组数据质控
Nanopore PromethION系统测序共得到6 988 795条raw reads, N50和平均读长分别为971 bp和881 bp,最大读长达到96 051 bp。raw reads的长度分布为1 kb到10 kb以上,其中分布reads数最多的长度是1 kb。此外,raw reads的Q值分布介于Q6~Q12,分布reads数最多的Q值为Q9。
2.2 全长转录本的鉴定和分析
经过滤得到6 953 469条clean reads,其中包含5 143 999条全长转录本。这些全长转录本的长度分布介于1~7 kb,分布在1 kb的转录本数量最多。进一步过滤冗余全长转录本,得到10 243条非冗余全长转录本,N50和平均读长分别为1 042 bp和894 bp,最大读长为4 855 bp;它们的长度分布介于1~5 kb,其中分布在1 kb长度的非冗余全长转录本数量最多。
2.3 全长转录本的数据库注释
分别有9 342, 4 038, 4 283, 2 569, 4 859和3 450条全长转录本可注释到Nr, KOG, eggNOG, Pfam, GO和KEGG数据库。注释转录本数量最多的物种是东方蜜蜂微孢子虫(8 700, 93.13%),其次是蜜蜂微孢子虫Nosemaapis(263, 2.82%)和家蚕微孢子虫Nosemabombycis(156, 1.67%)(图1: A)。注释到Nr数据库的全长转录本共有8 835条全长转录本注释为未知蛋白;注释到家蚕微孢子虫的156条全长转录本,与ATP催化与结合相关的全长转录本包括ONT.2029.1, ONT.1203.1, ONT.1993.3, ONT.77.4, ONT.972.4, ONT.972.6, ONT.972.5, ONT.972.1和ONT.972.3(图1: A)。东方蜜蜂微孢子虫转录本可注释到KOG数据库的25个功能分类,注释数量最多的是翻译、核糖体结构和生物合成(translation, ribosomal structure and biogenesis)(537, 13.30%),其次是翻译后修饰、蛋白质翻转和分子伴侣(posttranslational modification, protein turnover and chaperones)(490, 12.13%)以及转录(transcription)(452, 11.19%)(图1: B)。注释到eggNOG数据库的25个功能类别,注释数量最多的前3位分别是未知功能(function unknown)(903, 21.08%),翻译、核糖体结构和生物合成(623, 14.55%)以及复制、重组和修复(replication, recombination and repair)(538, 12.56%)(图1: C)。注释到GO数据库的43个功能条目,其中细胞组分(cellular component)大类中注释数量最多的是细胞(cell)(2 502, 51.49%)、细胞组件(cell part)(2 494, 51.33%)和细胞器(organelle)(1 923, 39.58%);分子功能(molecular function)大类中注释数量最多的是结合(binding)(2 431, 50.03%)、催化活性(catalytic activity)(2 212, 45.52%)和结构分子活性(structural molecule activity)(232, 4.77%);生物学进程(biological process)大类中注释数量最多的是细胞进程(cellular process)(2 795, 57.52%)、代谢进程(metabolic process)(2 603, 53.57%)和单一组织进程(single-organism process)(1 175, 24.18%)(图2: A)。注释到KEGG数据库的49条通路,其中注释数最多的通路是抗生素的生物合成(biosynthesis of antibiotics)(262, 7.59%)、氨基酸的生物合成(biosynthesis of amino acids)(138, 4.00%)、碳代谢(carbon metabolism)(126, 3.65%)、核糖体(ribosome)(112, 3.25%)和剪接体(spliceosome)(105, 3.04%)(图2: B)。
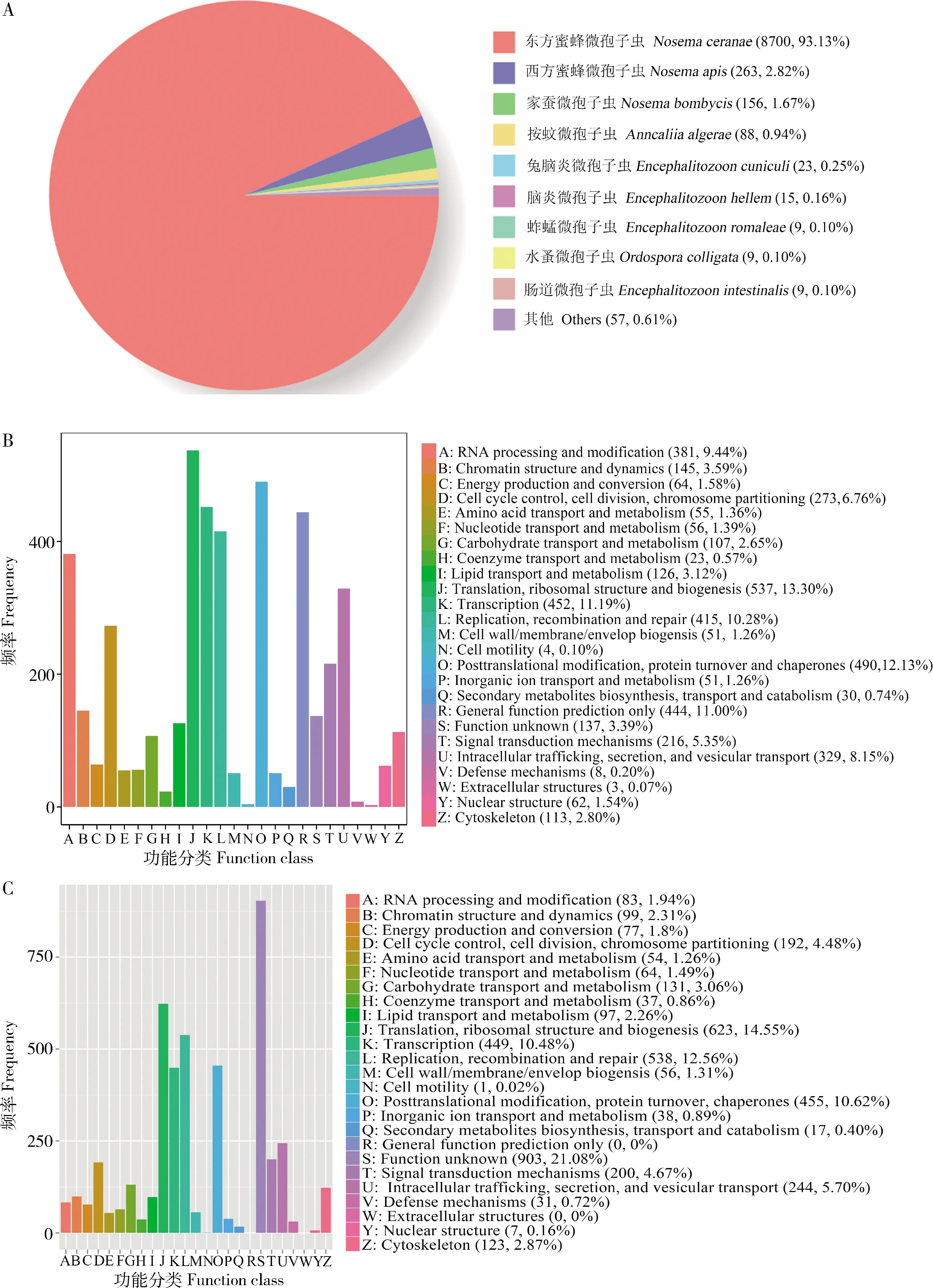
图1 东方蜜蜂微孢子虫全长转录本的Nr(A), KOG(B)和eggNOG(C)数据库注释
进一步分析发现,分别有6条和1条全长转录本可注释到Nr数据库中的ABC转运体相关编码基因和己糖激酶编码基因(表1);分别有2, 2和1条全长转录本可注释到孢壁蛋白编码基因、几丁质酶相关编码基因以及孢壁和锚定盘复合蛋白编码基因(表1)。
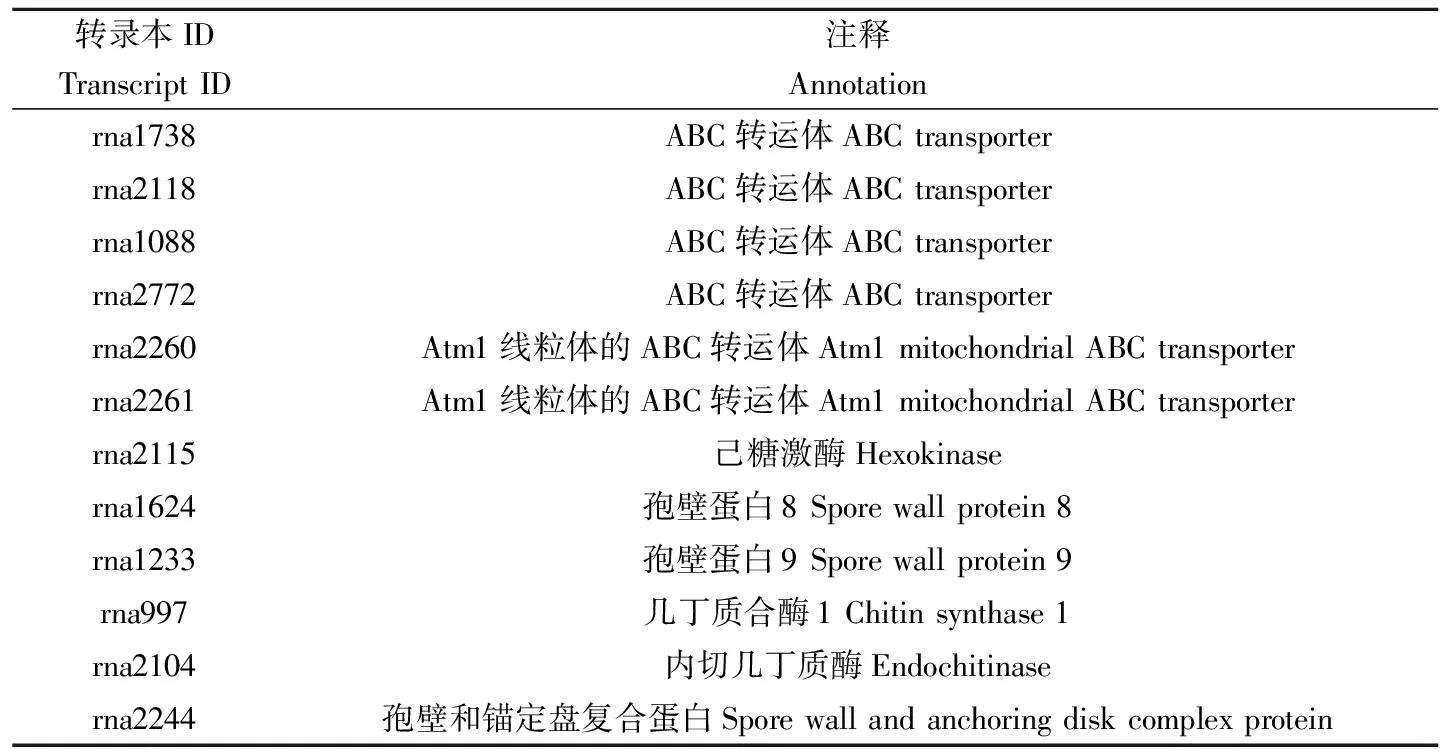
表1 东方蜜蜂微孢子虫全长转录本在Nr数据库中注释到的毒力和侵染相关因子
2.4 转录本中lncRNA的鉴定及分析
利用CNCI, CPC, Pfam和CPAT 4种方法分别鉴定出134, 301, 36和159个lncRNA,四者的交集为87个lncRNA(图3: A),为高可信度lncRNA;其中正义链lncRNA(sense lncRNA)、反义链lncRNA(anti-sense lncRNA)和基因间区lncRNA(intergenic lncRNA)的数量分别为49, 25和13个(图3: B)。
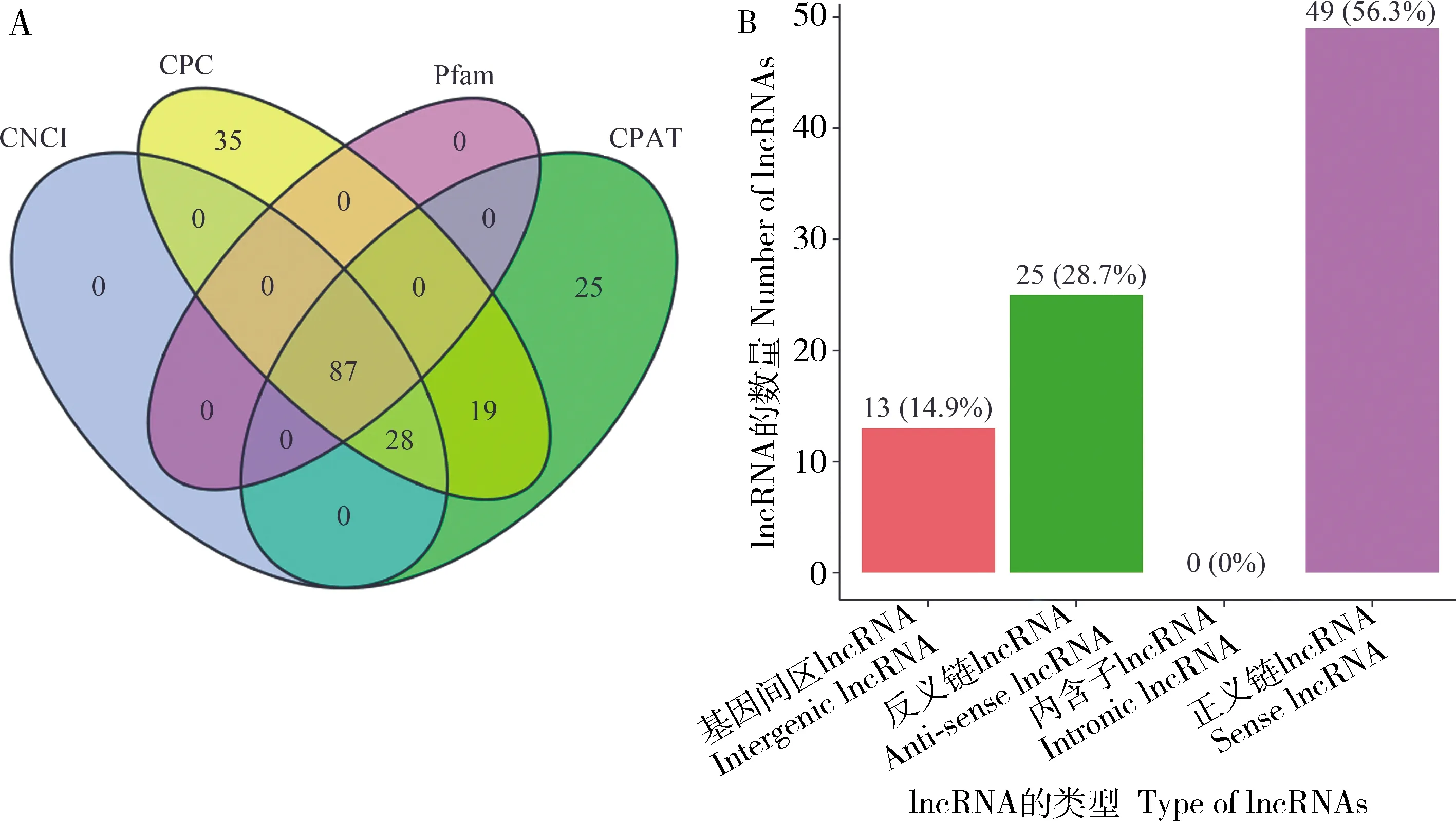
图3 东方蜜蜂微孢子虫转录组中lncRNA的数量和类型
2.5 全长转录本的表达量
测序饱和度分析结果显示,随着抽取的mapped reads数不断增多,检测到的表达的全长转录本数量趋于饱和(图4: A),说明本研究的测序量足以检测到全部表达的全长转录本。此外,全长转录本的表达量(CPM)范围在0.1到10 000以上,其中表达量在10附近的全长转录本丰度最高,约占所有转录本数量的45%(图4: B)。
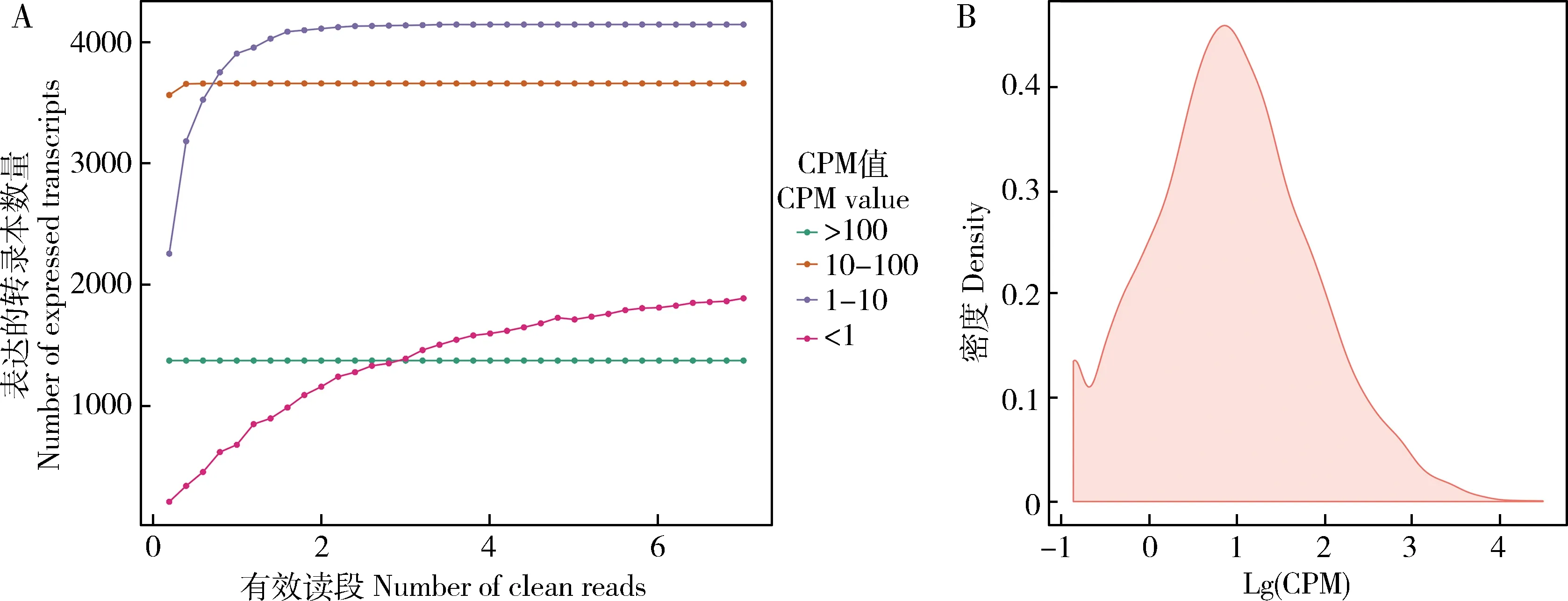
图4 东方蜜蜂微孢子虫转录组测序饱和度(A)和全长转录本的表达量分布(B)
3 讨论
第三代高通量测序技术因具有超长读段的特点,在鉴定全长转录本方面表现出独特优势。目前,东方蜜蜂微孢子虫的组学研究较为有限,高质量的参考转录组缺失。本研究利用Oxford Nanopore PromethION平台对东方蜜蜂微孢子虫的纯净孢子进行测序,获得了6 953 469条clean reads,鉴定到10 243条非冗余全长转录本。这些非冗余的全长转录本的N50和平均读长分别为1 042 bp和894 bp,最大读长达4 855 bp,体现出Nanopore测序技术在鉴定真菌全长转录本方面的优势。Workman等(2019)利用Nanopore技术对人类B淋巴细胞GM12878细胞系进行测序,鉴定到的全长转录本的N50和平均读长分别为1 334和771 bp,与本研究的结果类似。
进一步对上述全长转录本进行数据库注释,发现有9 342, 4 038, 4 283, 2 569, 4 859和3 450条全长转录本可分别注释到Nr, KOG, eggNOG, Pfam, GO和KEGG数据库。Nr数据库是NCBI中的非冗余蛋白质数据库,包含了Swiss-Prot, PIR(Protein Information Resource), PRF(Protein Research Foundation)和PDB(Protein Data Bank)蛋白质数据库以及从GenBank和RefSeq的CDS数据翻译过来的蛋白质数据信息。本研究中,共有多达93.13%的全长转录本注释到东方蜜蜂微孢子虫,与实际情况相符;另有2.82%和1.67%的全长转录本可分别注释到西方蜜蜂微孢子虫和家蚕微孢子虫(图1: A),体现了三者同属于Nosema属,亲缘关系较近。此外,进一步分析注释到家蚕微孢子虫的156条全长转录本,发现ONT.2029.1, ONT.1203.1, ONT.1993.3, ONT.77.4, ONT.972.4, ONT.972.6, ONT.972.5, ONT.972.1和ONT.972.3 9个全长转录本与ATP催化与结合相关,表明上述全长转录本在东方蜜蜂微孢子虫和家蚕微孢子虫侵染宿主过程参与对能量的获取与消耗等生命过程。长期以来,由于直接对微孢子虫进行转基因操作的技术体系缺失,导致微孢子虫基因功能研究举步维艰。本研究发现,共有8 835条全长转录本在Nr数据库中注释为未知蛋白,占全长转录本总数的86.25%,表明东方蜜蜂微孢子虫的多数基因功能仍有待于进一步深入研究。而基因功能的阐明首先需要对基因的全长序列进行克隆,本研究鉴定到的高质量全长转录本可为东方蜜蜂微孢子虫基因的全长序列克隆提供必要的数据基础。微孢子虫专营细胞内寄生,无法脱离宿主细胞而增殖(Gisder and Genersch, 2015)。到目前为止,来源于蜜蜂组织或器官的可稳定传代的体外培养细胞系仍然缺失,严重阻碍东方蜜蜂微孢子虫的基因功能研究。Gisder等(2011)研究证实东方蜜蜂微孢子虫和西方蜜蜂微孢子虫可通过极丝弹射的方式侵染鳞翅目舞毒蛾LymantriadisparIPL-LD-65Y细胞系,并在细胞内部稳定繁殖。Guo等(2016)曾通过碱液刺激家蚕微孢子虫孢子体外发芽,然后感染鳞翅目家蚕BmN细胞系,再利用脂质体包裹piggyBac转座子载体和pIZT/V5-His非转座子载体转染感染细胞,通过连续的荧光观察和分子生物学检测证实将二种载体携带的gfp基因成功导入家蚕微孢子虫基因组;同时,作者还通过饲喂家蚕微孢子虫孢子感染家蚕个体,继而注射脂质体包裹的上述两种载体,证实gfp基因成功插入家蚕微孢子虫的基因组。上述研究为后续开展东方蜜蜂微孢子虫的基因功能研究提供了思路和方法借鉴。
本研究发现,KOG数据库中注释全长转录本数量最多的功能分类是翻译、核糖体结构和生物合成(537, 13.30%),翻译后修饰、蛋白质翻转和分子伴侣(490,12.13%)以及转录(452, 11.19%);eggNOG数据库中注释全长转录本数量最多的是未知功能(903, 21.08%),翻译、核糖体结构和生物合成(623, 14.55%)以及复制、重组和修复(538, 12.56%)(图1:B, C)。本研究的测序材料为东方蜜蜂微孢子虫的纯净孢子,而孢子是病原的休眠态存在形式。上述结果表明病原孢子虫的孢子中仍然发生着遗传信息的传递及翻译后修饰等生命活动。本研究中,东方蜜蜂微孢子虫的全长转录本可注释到GO数据库中细胞组分、分子功能和生物学进程大类下的43个功能条目(图2: A),以及KEGG数据库中的49条通路,包括抗生素的生物合成、氨基酸的生物合成、碳代谢、核糖体、剪接体、RNA转运、嘌呤代谢、细胞周期、氧化磷酸化和嘧啶代谢等(图2: B)。以上注释结果为深入理解东方蜜蜂微孢子虫的基因功能和转录本功能提供了重要的参考信息。
东方蜜蜂微孢子虫的孢子壁主要由几丁质和孢壁蛋白组成,孢壁蛋白能够抵御外界的不良环境、传递外界环境变化信号和附着等作用(Southernetal., 2007)。由于缺少典型的线粒体,东方蜜蜂微孢子虫自身只能通过糖酵解途径产生少量ATP,大部分的ATP必须由宿主提供(Cornmanetal., 2009)。ABC转运蛋白的主要功能是结合ATP并利用ATP水解产生能量将物质逆浓度梯度运输(冯振月等, 2018)。本研究发现,6条东方蜜蜂微孢子虫的全长转录本可注释到与物质运输相关的ABC转运体相关蛋白编码基因,1条全长转录本可注释到糖酵解途径的关键酶己糖激酶编码基因,2条全长转录本可注释到孢壁蛋白编码基因,2条全长转录本注释到几丁质酶相关编码基因,1条全长转录本注释到孢壁和锚定盘复合蛋白编码基因(表1)。上述涉及毒力因子和侵染相关因子的全长转录本为深入理解东方蜜蜂微孢子虫的侵染机制提供了重要的参考信息。Nanopore测序技术的另一优势在于对不同剪接异构体进行精确定量(Magietal., 2018)。目前,我们已获得东方蜜蜂微孢子虫感染的意大利蜜蜂及中华蜜蜂工蜂中肠的全长转录组数据(未发表数据)。下一步将从上述包含宿主数据和病原数据的混合数据中筛滤出东方蜜蜂微孢子虫的纯净数据,结合本研究中东方蜜蜂微孢子虫的孢子转录组数据进行比较分析,进一步探究病原对蜜蜂宿主的侵染机制。
LncRNA已被证实在细胞周期(Tripathietal., 2013)、细胞分化(Lietal., 2016)、表观遗传(Kanduri, 2011)及剂量补偿效应(Gorodkin and Hofacker, 2011)等诸多生命活动中发挥重要的调控功能。然而,真菌的lncRNA研究进展缓慢,包括东方蜜蜂微孢子虫在内的蜜蜂病原的lncRNA研究极为滞后。前期研究中,笔者团队利用Illumina短读段测序技术对东方蜜蜂微孢子虫的纯净孢子进行测序,通过生物信息学方法首次鉴定出83个lncRNA,包括59条反义链lncRNA、21条lincRNA和3条正义链lncRNA(Guoetal., 2018b)。本研究中,我们基于病原孢子的全长转录组数据鉴定出87个lncRNA,包括49条正义链lncRNA、25条反义链lncRNA和13条基因间区lncRNA(图3)。将本研究中基于三代测序数据鉴定到的lncRNA序列与前期基于二代数据鉴定到的lncRNA序列进行比对,发现二者之间没有重叠。鉴于Nanopore测序读段与Illumina测序读段的差异,上述结果表明两种测序技术在鉴定lncRNA方面可以相互补充。本研究鉴定出的lncRNA进一步丰富了东方蜜蜂微孢子虫的lncRNA信息,为lncRNA的功能研究提供了必要基础,也为其他微孢子虫的lncRNA研究提供了有价值的参考信息。
人们对于真菌孢子中是否存在转录等生命活动认识较为有限(Williamsetal., 2005; Liuetal., 2016)。此前,笔者团队通过分子生物学和二代组学手段证实东方蜜蜂微孢子虫的孢子能够进行转录活动(Guoetal., 2018b)。本研究中,表达量计算结果显示东方蜜蜂微孢子虫的全长转录本的表达量(CPM)范围在0.1到10 000以上,约45%的全长转录本的表达量在10附近(图4)。前期研究中,笔者团队通过二代组学技术在东方蜜蜂微孢子虫的孢子中鉴定到83个表达的lncRNA(Guoetal., 2018b)和204个表达的circRNA(Guoetal., 2018a);此外还鉴定到11个表达的miRNA和2 076个表达的mRNA(未发表数据)。以上结果进一步证实,东方蜜蜂微孢子虫的孢子中的确存在转录活动。推测东方蜜蜂微孢子虫孢子提供基因转录形成物质和能量代谢相关的转录本,进而翻译形成相应的蛋白质或发挥转录和转录后水平的调控作用,以维持休眠态孢子必要的新陈代谢。但真菌孢子中存在转录活动是否为普遍现象,仍需要进一步研究。
综上所述,本研究首次利用Nanopore技术构建和注释东方蜜蜂微孢子虫的全长转录组,高质量的全长转录组可为东方蜜蜂微孢子虫的比较转录组分析,转录本的可变剪接和可变腺苷酸化分析,SSR位点挖掘,基因结构优化,以及基因全长序列克隆及功能研究提供关键基础。
