Bile acid synthetic defects: Simplified approach in a nutshell Aathira Ravindranath, Moinak Sen Sarma *, Surender Kumar Yachha
2020-03-03DepartmentofPediatricGastroenterologySanjayGandhiPostGraduateInstituteofMedicalSciencesLucknowUttarPradeshIndia
Department of Pediatric Gastroenterology, Sanjay Gandhi Post Graduate Institute of Medical Sciences, Lucknow, Uttar Pradesh, India
Bile acid synthetic defects (BASD) constitute a rare group of dis- orders with manifestations in children as well as adults. Diagnos- ing the individual BASD mandates a lucid understanding of the bile acid synthetic pathway. However, the synthesis pathway is com- plex, consisting of numerous enzymes and intermediate products that are volatile in the physician’s memory. Despite paucity of lit- erature, a simplified approach to the BASD is presented in this paper.
There are two major pathways of bile acid synthesis in human beings: classical and alternate (acidic) pathways. Cholesterol is the common precursor of both pathways. After undergoing multiple complex steps, classical and alternate pathways finally produce primary bile acids - cholic acid (CA) and chenodeoxycholic acid (CDCA), respectively. The alternate pathway predominantly func- tions in the fetus whereas the classical pathway takes lead after birth and persists throughout adulthood. While the classical path- way works on the sterol ring first, the alternate pathway targets primarily the side chain of cholesterol. The intermediary substrates formed in the classical pathway undergo further side-chain oxida- tion in the peroxisomes. A simplified bile acid synthesis pathway with the clinically relevant enzymes is shown in Fig. 1 . Deficien- cies of these enzymes at each step will produce different clinical manifestations.
CA and CDCA are nontoxic bile acids which are polar, water soluble and have excellent cholerectic and surfactant properties. Mono and di-hydroxy bile acids are normally formed during the process of bile acid synthesis but they are rapidly converted to nontoxic forms. However, in cases of BASD these atypical bile acids accumulate which are toxic to hepatocytes and they also inhibit canalicular transport of bile acids. Normally, CA and CDCA exert negative feedback by activating farnesoid X receptor (FXR) and small heterodimer partner (SHP). These in turn inhibit the gene that encodes the rate limiting enzyme cholesterol 7 α-hydroxylase thereby regulating bile acid production [1] . After production of CA and CDCA, bile acid ligase and transferase form glycine and taurine conjugates which are then transported into the bile canaliculus and thereafter to the small intestine where bacteria deconjugate a small fraction of bile acids which are passively absorbed and re- turned to the liver. Bacteria also cause epimerization of CDCA and convert it to ursodeoxycholic acid (UDCA) which then becomes a part of the bile acid pool. UDCA is commonly used in cholestatic disorders and it will affect the composition of bile acid pool by re- ducing CA and CDCA levels and the predominant bile acid would be UDCA [2] . After reaching the ileum, 90% bile acids undergo en- terohepatic circulation thereby conserving the bile acid pool and 10% reach the colon where secondary bile acids lithocholic and de- oxycholic acids are formed from CDCA and CA, respectively. Entero- hepatic circulation is maintained by action of apical sodium bile acid transporter (ASBT) which actively transports luminal bile acids into the enterocyte. Organic solute transporter α/ β (OST α/ β) is present in the basolateral membrane and transports bile acids into portal blood from enterocyte ( Fig. 2 ) [3] . Functions of the bile acids include formation of micelles in gut lumen, absorption of fat and fat soluble vitamins, cholesterol homeostasis, induction of bile flow through feedback mechanism, glucose homeostasis and antimicro- bial activity.
Congenital deficiencies in the crucial enzymes involved in bile acid synthetic pathway result in several disorders grouped together as BASD. The effect of BASD can be broadly conceived to result from accumulation of toxic intermediaries which cause liver injury and absence of bile acids that causes steatorrhea and fat soluble vitamin deficiencies. Clinical features of BASD have con- siderable phenotypic overlap with progressive familial intrahepatic cholestasis (PFIC) and peroxisomal disorders. Once the bile acids have formed, defects at bile acid uptake, excretion or transporter levels due to genetic mutation in the proteins is termed as PFIC. It is well understood that PFIC will have intense pruritus due to ac- cumulated bile acids stimulating pruritogens while BASD will have paucity of pruritus. Since there is deficiency of bile acids entering the biliary canaliculi, both BASD and PFIC will have low-normal gamma-glutamyl transpeptidase (GGT). For simplicity, peroxisomal disorders can be viewed as “extended” BASD due to deficiency in the oxidation of intermediary substrates that enter the peroxisome. Of all BASD, 3 β-hydroxysteroid oxidoreductase and 3-oxosteroid 5 β-reductase deficiencies are the most frequent single enzyme defects of bile acid synthesis pathway [4] . Since classical pathway is the main pathway that produces 80% -90% of the bile acids after birth, its defects are more pronounced in infancy and child- hood. Alternate pathway defects are less common and will be manifested either in utero or in adulthood with gradual accu- mulation of the toxic intermediates [5] . In the classical pathway, defect at each progressive step leads to more severe and diverse manifestations due to the toxic effects of the accumulated interme- diates. Defects of the alternate pathway present with multi-organ involvement that may or may not be associated with cholestasis. Table 1 is a summary of the clinically relevant defects.
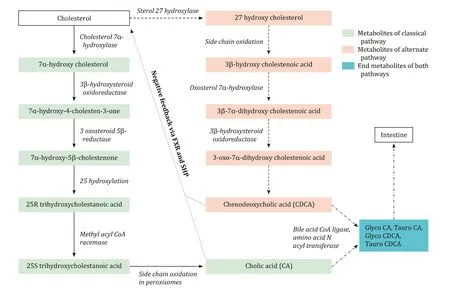
Fig. 1. Simplified bile acid synthetic pathway. Solid arrows: classical pathway; dashed arrows: alternate pathway; dotted arrows: negative feedback; dashed and dotted arrows: common steps. Enzymes of classical and alternate pathway are shown in italics.
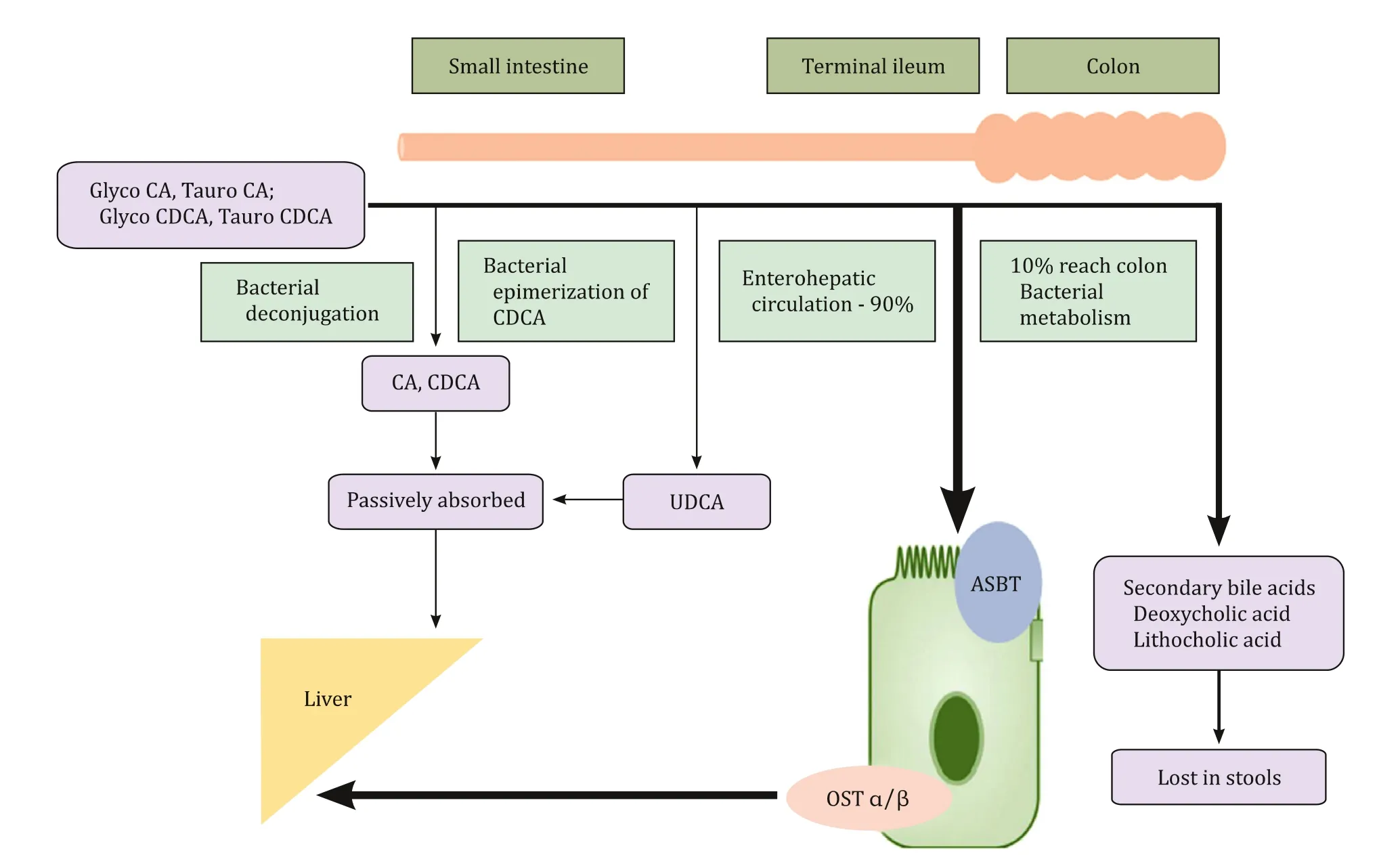
Fig. 2. Circulation of bile acids. CA: cholic acid; CDCA: chenodeoxycholic acid; UDCA: ursodeoxycholic acid; ASBT: apical sodium bile acid transporter; OST α/ β: organic solute transporter α/ β.
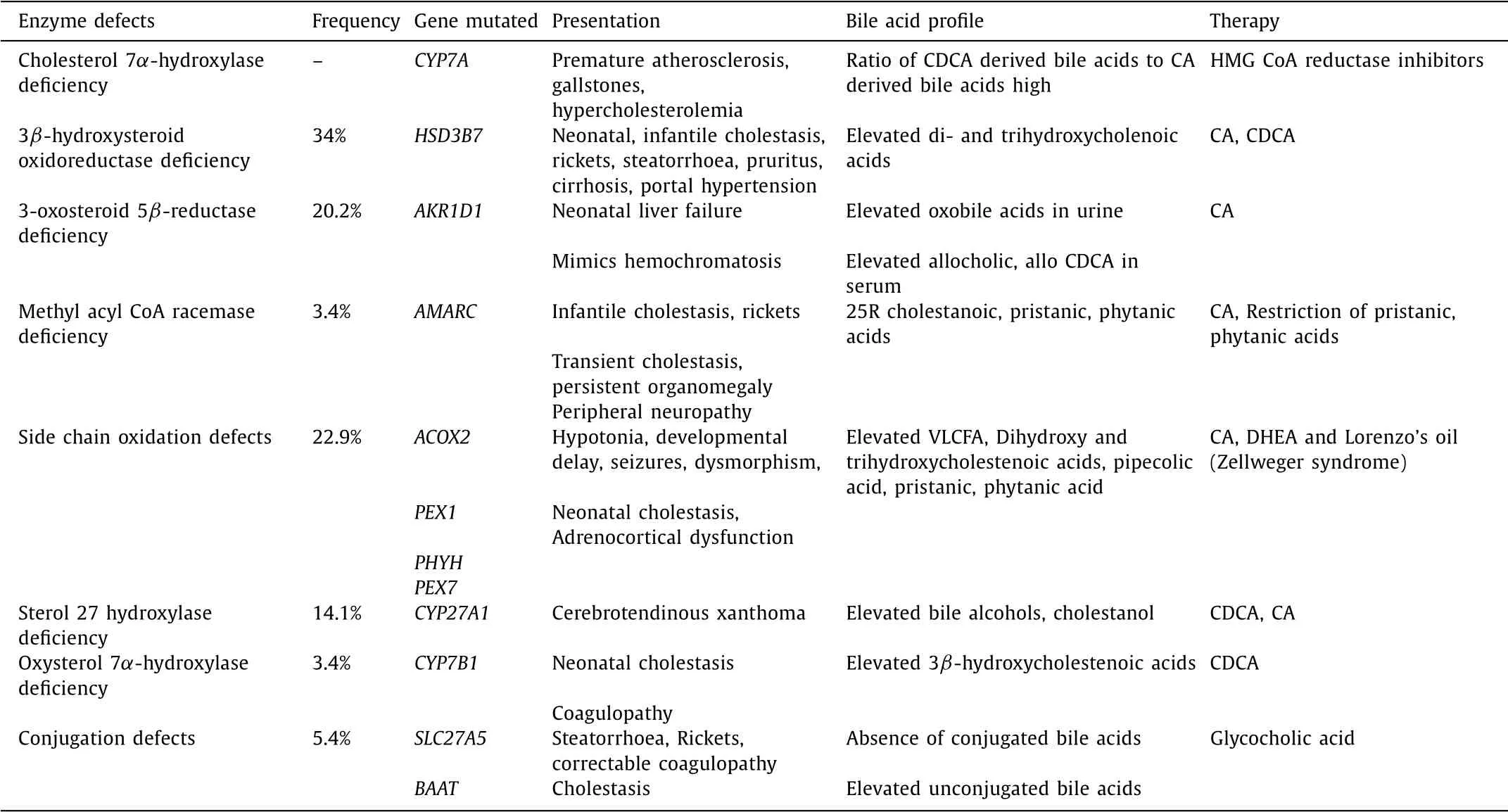
Table 1 Summary of clinical and biochemical features of bile acid synthetic defects.
As BASDs are evolving disorders, determining the exact preva- lence is difficult. Studies from Saudi Arabia and Japan showed that BASD accounted for 2% -6.3% of all liver diseases that in- cluded infantile cholestasis, cirrhosis, chronic hepatitis, liver fail- ure and vitamin D resistant rickets [ 6 , 7 ]. The combined incidence of 3 β-hydroxysteroid oxidoreductase deficiency and 3-oxosteroid 5 β-reductase deficiency in a European survey was 1.13 per 10 million [8] . Experience from Cincinnati with 13,800 mass spec- troscopy screenings also suggests that BASD accounts for 2% of all cholestatic liver diseases [4] .
Cholesterol 7 α-hydroxylase is the first enzyme in the classi- cal pathway and assumes importance for being the rate limit- ing enzyme. Defect in this enzyme due to homozygous muta- tion in CYP7A gene will result in accumulation of cholesterol. The cholesterol is hence easily and preferably shunted to the alter- nate pathway for bile acid synthesis. As no atypical intermedi- aries are formed, there will be no liver injury. The only manifes- tations of this disorder would be hypercholesterolemia, premature atherosclerosis and gallstones [9] .
3 β-hydroxysteroid oxidoreductase catalyses conversion of 7 αhydroxy cholesterol to 7 α-hydroxy-4-cholesten-3-one and its de- fect results in accumulation of 7 α-hydroxy cholesterol which undergoes side chain oxidation and hydroxylation to form di- hydroxy and trihydroxycholenoic acids. Mutations in HSD3B7 gene inherited in an autosomal recessive manner results in 3 β- hydroxysteroid oxidoreductase deficiency [10] . It is the commonest BASD and has the most heterogenous clinical presentation ranging from cholestasis in children to cirrhosis in adults [ 11 , 12 ].
In a series of 14 symptomatic patients with 3 β-hydroxysteroid oxidoreductase defect, the median age of presentation was 8 (range 2-36) weeks. Presenting features were persistent neonatal cholestasis ( n = 11), rickets ( n = 8), pruritus ( n = 3) and steatorrhoea ( n = 3). Cirrhosis and portal hypertension was also documented in a 2-year-old child. All patients had normal GGT. Vitamin D and E deficiencies were documented in the majority. Liver histology showed non-specific changes such as extensive giant cell transfor- mation, hepatocellular disarray, extramedullary hematopoiesis and cholestasis. Fast atom bombardment-mass spectrometry (FAB-MS) or electrospray tandem mass spectrometry revealed elevated di- and trihydroxycholenoic acids. Treatment with CA (4-8 mg/kg/d) or CDCA (7-18 mg/kg/d) lead to symptom resolution, normalization of biochemistry and even reversal of abnormal liver histology in 12/13 children [13] .
3-oxosteroid 5 β-reductase converts 7 α-hydroxy-4-cholesten-3- one to 7 α-hydroxy-5 β-cholestenone. Deficiency of this enzyme re- sults from homozygous or compound heterozygous mutations in AKR1D1 gene on chromosome 7q33 and causes increase in serum allo CA, allo CDCA and oxo bile acids. This disorder presents as severe liver injury with progressive neonatal cholestasis, coagu- lopathy and rapid liver dysfunction if untreated. Since advanced neonatal liver disease has impaired iron handling, this disorder can closely mimic neonatal hemochromatosis with elevated serum ferritin and extrahepatic iron deposition [14] . For reasons un- known, few cases have reported elevated GGT in 3-oxosteroid 5 β- reductase deficiency [15] . Liver biopsy shows lobular disarray, giant cell transformation and slit-like bile ducts on electron microscopy. Though there are sporadic reports of improvement with UDCA, treatment of choice is cholic acid which has to be initiated early for optimal outcome [15] .
Methylacyl-CoA racemase converts 25R trihydroxycholestanoic acid to its 25S enantiomer. It also plays a crucial role in branched chain fatty acid metabolism by converting 25R pristanoyl CoA to 25S enantiomer. Therefore, defect in this enzyme causes problems in bile acid as well as fatty acid metabolism. This disease may present at any age. Predominant presentation in infants is cholesta- sis, rickets, coagulopathy; in children transient cholestasis and per- sistent organomegaly; and in adulthood peripheral neuropathy due to pristanic and phytanic acid accumulation [16] . FAB-MS reveals elevated 25R cholestanoic, pristanic, phytanic acids. It can be con- firmed by detecting homozygous mutations in AMARC gene on chromosome 5p13. Treatment with CA reverses liver injury. Dietary restriction of phytanic and pristanic acids are required to prevent neurotoxicity by avoiding cream, butter and fish oil [17] .
Disorders affecting side chain oxidation can stem either from single enzyme defects or peroxisomal biogenesis defects which in- clude Zellweger syndrome, Refsum disease and adrenoleukodys- trophy. In humans, Acyl CoA oxidase (mutation in ACOX2 gene) and trihydroxycholestanoic acid CoA oxidase deficiencies are the most frequently described single enzyme oxidation defects. Defec- tive oxidation affects bile acids and also very-long-chain fatty acids (VLCFA). Long-chain fatty acids are required for myelination and hence, neurological manifestations are more overwhelming than cholestasis, coagulopathy and organomegaly in these patients [18] .
The distinct syndrome of cerebrotendinous xanthoma (CTX) is caused by defect in the first enzyme of the alternate pathway, sterol 27 hydroxylase enocoded by CYP27A1 gene on chromo- some 2q35. Defective CDCA production releases the feedback in- hibition on 7 α- hydroxylase. As a result there will be low choles- terol, increase in cholestanol and bile alcohol glucoronides. CTX can present in the neonatal period with cholestasis, during infancy with diarrhea, in adolescents with cataract and in adults with neu- rologic symptoms and xanthomata [19] . CDCA replacement therapy may cause deterioration in 60% patients [20] .
The importance of the alternate pathway in the fetus and early neonatal period is reflected by defect in oxysterol 7 α-hydroxylase that causes cholestasis, coagulopathy and progressive liver dysfunc- tion soon after birth. It is an infrequently reported disorder due to mutation in CYP7B1 gene. FAB-MS will indicate elevated 3 βhydroxy-5-cholenoic and 3 β-hydroxy-5-cholestenoic acids. It has poor prognosis but early CDCA replacement may be effective [21] .
Bile acid CoA ligase (encoded by SLC27A5) and bile acid-CoA: amino acid N-acetyl transferase (encoded by BAAT) act sequen- tially to produce glycine and taurine conjugates of bile acids. The conjugated bile acids are ionized and are solely absorbed by ac- tive ileal transport. When there are defects in conjugation, uncon- jugated bile acids reaching the intestine are rapidly absorbed by passive diffusion and are unable to form micelles, thus causing malabsorption of fat and fat soluble vitamins. The serum level of unconjugated bile acids will be elevated [22] . These children can present with steatorrhoea and fat soluble vitamin deficiencies. In a series of 10 patients, 5 had rickets, 4 had coagulopathy and 4 had cholestasis. Oral glycocholic acid and fat soluble vitamin sup- plementation can alleviate symptoms [23] .
A physician should consider BASD in the following situations: (i) neonatal cholestasis without pruritus and low-normal GGT; (ii) cholestasis in infancy (persistent or transient) with predominant rickets and/or steatorrhea; (iii) neonatal liver failure with low- normal GGT; (iv) cryptogenic cirrhosis with history of transient neonatal cholestasis; (v) overwhelming neurological presentation with cholestasis. Presence of pruritus and high GGT are atypical presentations.
Liver biopsy is not discriminatory or pathognomonic for any of the BASD though cholangiocentric hepatitis can be seen in 3 β- hydroxysteroid oxidoreductase deficiency. Serum and urinary bile acid measurement are screening tests. Enzymatic, colorimetric, and fluorometric assays determine total primary bile acids which are expected to be low except in conjugation defects where serum un- conjugated bile acid level is high. In contrast PFIC has elevated total bile acids. FAB-MS uncovers the profile of all bile acid in- termediates that indicate which enzyme is blocked. FAB-MS is the quickest screening tool but needs considerable expertise in anal- ysis. Therapeutic UDCA supplementation ought to be stopped at least 5 days before FAB-MS as it will interfere with bile acid metabolite determination. Enzyme and mutational analysis are confirmatory investigations. Thus, diagnostic approach will entail demonstration of low serum primary bile acids followed by de- tection of abnormal bile acid metabolites in urine or serum by FAB-MS and confirmed by genetic studies or enzymatic assay in fibroblasts.
Diagnosing BASD assumes utmost significance because of the possibility of reversing the disease process with bile acid replace- ment therapy. UDCA which is commonly used in cholestatic disor- ders may work by bile acid displacement. However it is not thera- peutic in BASD as it will not be able to generate the primary bile acids or reduce toxic bile acid production to a great extent. Cholic acid and CDCA are the mainstay of therapy in almost all the en- zymatic defects. They exert feedback inhibition on 7 α-hydroxylase which is the rate limiting step of bile acid synthesis, thus stalling generation of toxic bile acid intermediates. They also induce bile flow. UDCA should never be used concomitantly with CA as UDCA will inhibit ileal absorption of CA. Conjugation disorders require re- placement of glycocholic acid which is pre-conjugated. Early initia- tion of treatment can reverse not only biochemical parameters but also histopathological changes in liver [24] . Liver functions ought to be monitored 6 monthly and compliance to therapy has to be assessed by urine FAB-MS. Screening for hepatocellular carcinoma by serum alpha-fetoprotein and abdominal ultrasonography is rec- ommended [4] .
In conclusion, BASDs are rare disorders that cause liver dys- function, fat and fat soluble vitamin malabsorption. Though these are infrequently encountered, awareness about these disorders is essential because they are potentially treatable conditions if diag- nosed early.
CRediT authorship contribution statement
Aathira Ravindranath:Data curation, Writing - original draft.Moinak Sen Sarma:Conceptualization, Supervision, Writing - re- view & editing.Surender Kumar Yachha:Supervision, Writing - review & editing.
Funding
None.
Ethical approval
Not needed.
Competing interest
No benefits in any form have been received or will be received from a commercial party related directly or indirectly to the sub- ject of this article.
杂志排行

Hepatobiliary & Pancreatic Diseases International的其它文章
- Impact of EBV infection and immune function assay for lymphoproliferative disorder in pediatric patients after liver transplantation: A single-center experience
- Hepatobiliary&Pancreatic Diseases International
- Intraoperative management and early post-operative outcomes of patients with coronary artery disease who underwent orthotopic liver transplantation
- Acute onset of autoimmune hepatitis in children and adolescents
- Liver stiffness as a predictor of hepatocellular carcinoma behavior in patients with hepatitis C related liver cirrhosis ✩
- Treatment and prognosis of hepatic epithelioid hemangioendothelioma based on SEER data analysis from 1973 to 2014