介导巨噬细胞极化的关键转录因子及其与TLRs信号的相关性①
2020-02-29常晓彤
马 玉 郭 豪 常晓彤
(河北北方学院临床检验诊断学重点实验室,张家口 075000)
巨噬细胞是天然免疫系统中的一个重要成员,其来源于骨髓的造血干细胞,后者首先在外周血中分化为成熟的单核细胞,再转运到机体的大部分组织中,发育为巨噬细胞。基于巨噬细胞的可塑性和激活方式的不同[1],可将其分为经典活化型巨噬细胞(classical activated macrophage,即M1型)和选择性活化型巨噬细胞(alternative activated macrophage,即M2 型)[2],M1为促炎表型,M2为抗炎表型,当应对不同的微环境信号时,巨噬细胞可在M1表型和M2表型间迅速转变[3],以发挥其促炎或抗炎作用。巨噬细胞的极化过程依赖于许多关键转录因子的调控,在不同疾病中,相关转录因子可直接或间接作用于目标基因的调控区域,促进促炎或抗炎细胞因子的表达,驱使巨噬细胞功能表型极化。
巨噬细胞对非己成分的识别是固有免疫应答的重要事件。巨噬细胞可表达多种模式识别受体(pattern recognition receptors,PRRs),其中Toll样受体(toll-like receptors,TLRs)是重要的一类。TLRs可识别病原生物的病原相关分子模式(pathogen associated molecular patterns,PAMPs)[4]和内源性的危险相关分子模式(danger-associated molecular patterns,DAMPs)[5]。目前,在哺乳动物中,共发现13种TLRs,其中已确认的人类TLRs家族成员有10个,即TLR1~TLR10[2]。巨噬细胞的TLRs通过识别非已成分,启动其信号转导通路,参与病原体的清除或在无菌炎症下修复受损的细胞和基质以维护组织完整性及其功能[6]。调控巨噬细胞极化的关键转录因子与TLRs通路存在交互作用,影响巨噬细胞的极化过程和表型。本文对此作一综述,以期为TLRs和巨噬细胞的相关研究提供理论参考。
1 巨噬细胞极化
巨噬细胞功能广泛,在应答各种环境或病理环境变化时,巨噬细胞可经历不同表型的极化,获得不同的功能表型。M1型巨噬细胞由粒细胞-巨噬细胞集落刺激因子(granulocyte-macrophage colonystim-ulating factor,GM-CSF)、脂多糖(lipopolysacch-aride,LPS)、干扰素-γ(interferon-γ,IFN-γ)等诱导激活,可分泌肿瘤坏死因子-α(tumor necrosis factor-α,TNF-α)、IL-1、IL-6、活性氮和活性氧中间体等促炎因子和趋化因子,通过其分泌,激活适应性免疫应答,发挥专业抗原提呈细胞的作用,杀灭病原微生物,破坏肿瘤恶性细胞等[7]。M2型巨噬细胞可由巨噬细胞集落刺激因子(macrophage colony-stimulating factor,M-CSF)诱导活化。根据刺激物的不同, M2型巨噬细胞可进一步分为不同的亚型:在IL-4、IL-13的刺激下,可诱导成M2a型巨噬细胞,分泌精氨酸酶1 (Arginase1,Arg 1)和IL-10;在免疫复合物和 IL-1受体的配体等刺激下可诱导成M2b型巨噬细胞,其特点是高表达IL-10,低表达IL-12、IL-1β 和 TNF-α;在IL-10、转化生长因子-β(transforming growth factor-β,TGF-β)或糖皮质激素的激活下可诱导成M2c型巨噬细胞,可分泌IL-10、甘露糖受体(CD206)、TGF-β等[8]。M2型巨噬细胞具有较低的抗原呈递能力和抗炎作用,可下调适应性免疫应答,与寄生虫感染、组织重塑和肿瘤进展有关[9]。
2 介导巨噬细胞极化的关键转录因子及与TLRs信号的关系
既有研究表明,巨噬细胞极化依赖于转录因子的介导[10],如信号转导子和转录激活因子(signal transducer and activator of transcription,STATs)、干扰素调节因子(interferon-regulatoryfactor,IRFs)、核因子-κB(nuclear factor,NF-κB)、激活蛋白1(activator protein 1,AP-1)、过氧化物酶体增殖物激活受体-γ(peroxisome proliferator-activated receptor-γ,PPAR-γ)、cAMP反应元件结合蛋白(cAMP-responsive element-binding protein,CREB)、Krüppel样因子4(krüppel-like factor 4,KLF4)等,这些关键的转录因子的激活,控制着巨噬细胞M1-M2的极化方向,使向M1表型倾斜或向M2表型倾斜[11]。在巨噬细胞极化过程中,TLRS对极化调节发挥着重要的作用。
2.1STATs STATs是一组广泛表达于哺乳动物不同类型细胞和组织中的、能够与目的基因调控区DNA结合的转录因子,目前共发现8种亚型[12]。活化的STATs参与巨噬细胞极化,而STATs的活化依赖于一类可溶性的酪氨酸蛋白激酶(Janus kinases,JAKs),形成JAK/STAT信号通路。在巨噬细胞极化过程中,巨噬细胞的活化因子可导致受体相关的JAKs激活,STAT在JAK作用下发生酪氨酸磷酸化并二聚化,二聚化的STAT转移到细胞核内,结合目的基因的启动子序列并激活相应基因的转录[13]。JAK/STAT信号通路通过对应的细胞因子受体传递来自IFN-γ、IL-4、IL-10、IL-13等活化因子的信号,活化不同的STATs,产生不同的生物学应答。IFN-γ与巨噬细胞表面IFN-γ受体(IFN-γR)结合激活STAT1,导致促炎细胞因子的产生,使巨噬细胞倾向M1表型。而IL-4、IL-10和IL-13经JAK/STAT通路,使巨噬细胞倾向M2表型,IL-4与巨噬细胞表面的IL-4受体(IL-4R)结合激活STAT3和STAT6;IL-10与巨噬细胞表面的IL-10受体(IL-10R)结合激活STAT3;IL-13与巨噬细胞表面的IL-13受体(IL-13R)结合既能活化STAT3和STAT6,也能活化STAT1,但前者处于优势状态,因此,IL-4、IL-10和IL-13使巨噬细胞倾向M2型[9]。
在分子水平,STAT1和STAT3/6活化的平衡,紧密调控着巨噬细胞的极化和功能,经IL-4诱导STAT6活化的巨噬细胞可抑制经IFN-γ诱导STAT1活化的巨噬细胞的极化。TLRs信号通路与JAK/STAT通路存在交叉对话,两者相互作用,影响M1-M2巨噬细胞极化。巨噬细胞经IFN-γ极化后,对TLR4配体的促炎症应答增强,表明IFN-γ/JAK/STAT通路与TLR4通路在STAT1水平上存在汇集[14]。已知在大多数丙型肝炎患者中,丙型肝炎病毒(hepatitis C virus,HCV)持续感染并难以清除,有研究发现造成该结果的一个重要原因是HCV核心蛋白抑制了单核细胞向巨噬细胞的极化,其分子机制是HCV核心蛋白通过活化TLR2后,抑制了下游信号分子STAT1和STAT3的磷酸化,从而M1型巨噬细胞和M2型巨噬细胞的极化均受到抑制,导致M1和M2巨噬细胞功能紊乱,吞噬活性降低[15](图1)。
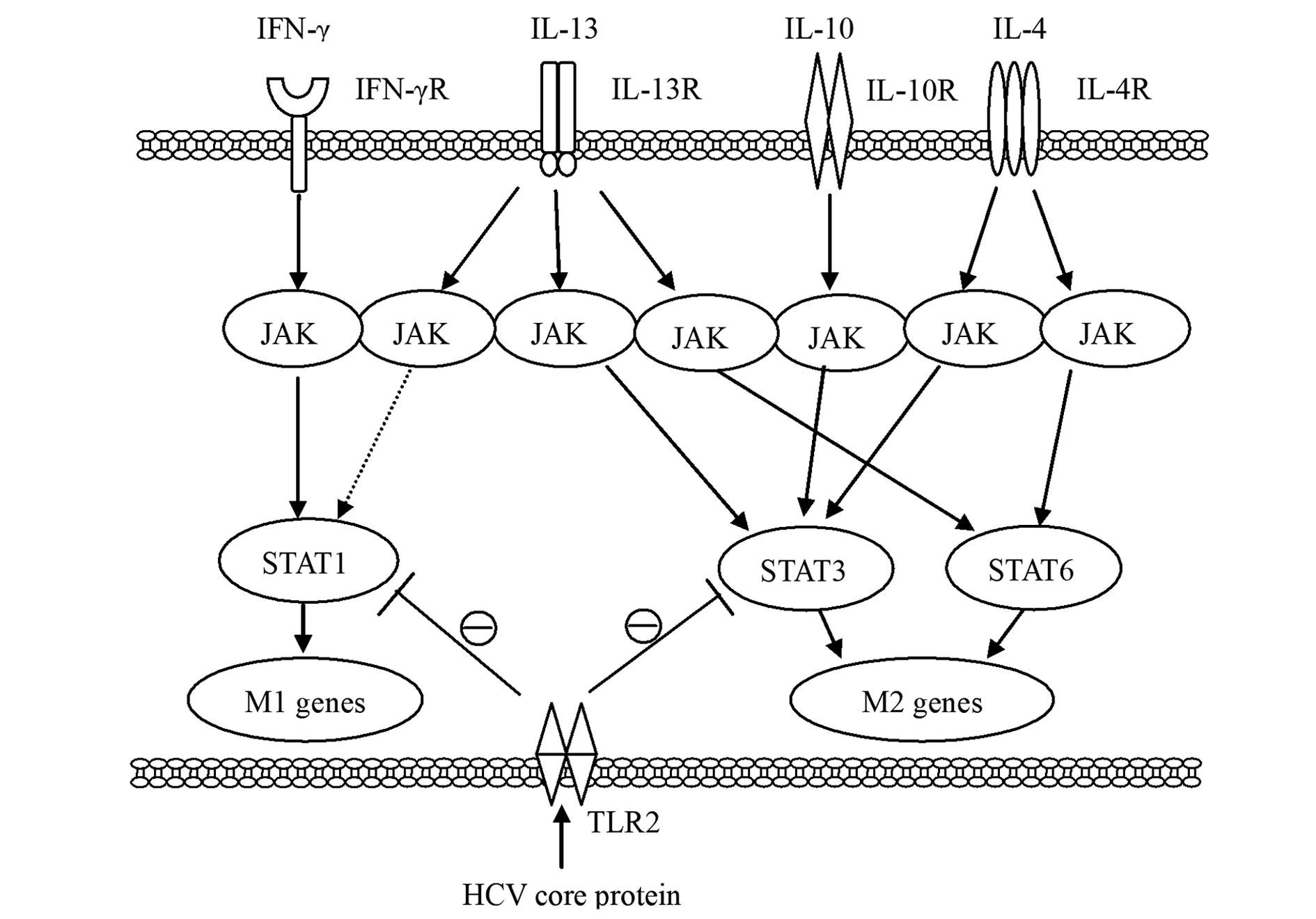
图1 STATs对巨噬细胞极化的影响及与TLRs的相关性Fig.1 Effect of STATs on macrophage polarization and correlation with TLRs
2.2IRFs IRFs是一类对IFN基因表达起调控作用的转录因子,能与Ⅰ型干扰素启动子区域的病毒应答元件结合,激活IFN-α的表达,目前在哺乳动物中共发现9种亚型。IRFs的N-端区有保守的DNA结合域,它们具有显著的同源性,在C-端区除了IRF6外均有IRF关联域(IRF-association domains,IADs),可与其他亚型的IRF相互作用。巨噬细胞的极化受IRFs的影响,其中IRF1、IRF5和IRF8主要参与促炎M1型巨噬细胞的极化,IRF3和IRF4主要参与抗炎M2型巨噬细胞的极化[16]。IRF4和IRF5除上述结构外,在IAD1和C-末端之间还有调控域和抑制区,被磷酸化激活时,可将抑制区转化为活化区,参与巨噬细胞极化反应[17]。TLRs信号通路影响IRFs介导的巨噬细胞极化,发现经IFN-γ极化的巨噬细胞,在应答TLR4配体的刺激时,IRF8表达增高,促炎性细胞因子表达进一步增强,表明IFN-γ通路和TLR4信号通路在IRF8水平汇集,促进M1巨噬细胞表型[18]。IRF4与TLRs的一个重要接合子——髓样分化因子(myeloid differentiation factor 88,MyD88)结合后,对TLRs信号的促炎症活性起到负调节的作用,而IRF5则可增强依赖TLRs诱导的TNF-α、IL-6、IL-12等细胞因子的产生,在巨噬细胞极化中,IRF4与IRF5竞争结合MyD88,向NF-κB、AP-1和其他炎症因子传递TLRs的不同激活信号,从而使促炎和抗炎达到平衡状态[19](图2)。
2.3NF-κB 在无活性的巨噬细胞内,核转录因子NF-κB与其抑制蛋白IκB结合,以无活性的异二聚体的形式存在于胞质内。当IκB被磷酸化后,NF-κB去抑制,游离的NF-κB转移至细胞核内,调控炎症、免疫应答等相关基因的表达。TLRs可识别特定的配体,除了TLR3外的TLRs均可导致NF-κB的激活[20]。细胞因子或PAMPs/DAMPs通过结合细胞表面TLRs,启动NF-κB介导的信号级联反应[1,21]。当巨噬细胞TLRs应答特定配体的刺激时,在多数情况下,NF-κB活化后,上调促炎症基因的表达,使巨噬细胞倾向M1表型。但由于产生的NF-κB亚基组成存在两种形式,即促炎症反应的p65/p50 M1型和抗炎症反应的p50/p50 M2型,当NF-κB以p65/p50的形式活化时,促炎细胞因子产生增加,形成M1表型;如果NF-κB以p50/p50的形式活化时,抗炎细胞因子产生增加,形成M2表型。所以,TLRs在应答LPS等的刺激时,能够转换巨噬细胞表型,产生M2型细胞因子,这是调节巨噬细胞可塑性的机制之一,有利于对感染和微环境的变化迅速作出应答[1,22](图3)。

图3 NF-κB对巨噬细胞极化的影响及与TLRs的相关性Fig.3 Effect of NF-κB on macrophage polarization and correlation with TLRs
2.4AP-1 在哺乳动物细胞中,转录因子激活蛋白AP-1主要是由c-Jun和c-Fos组成的同源c-Jun/c-Jun或异源c-Jun/c-Fos二聚体复合物,是多种细胞信号传导途径在细胞核内的交汇点,能够调控诸多与细胞分裂、增殖、炎症等相关基因的转录,是细胞增殖、分化、凋亡的关键决定因素之一。
在巨噬细胞内,TLRs或IL-1受体通过偶联MyD88,启动MAPKs信号级联放大反应,激活巨噬细胞中的AP-1,进而AP-1通过激活细胞因子的产生来调节炎症反应,TLRs或IL-1受体/MyD88/MAPKs/AP-1通路是巨噬细胞调节炎症过程的一条重要通路;c-Jun因子是构成AP-1的核心成分,比c-Fos因子,对巨噬细胞的活化更为关键[23]。AP-1受磷酸化调控,负责c-Jun磷酸化的酶是c-Jun N端激酶(c-Jun N-terminal kinase,JNK),JNK是MAPKs家族成员之一,当细胞受到不同的刺激(如Th1或Th2细胞因子的刺激),JNK磷酸化c-Jun的不同位点,发挥正向或负向调节作用,促进或抑制抗炎基因的表达。在促炎因素的诱导下,AP-1诱导促炎基因的表达,有研究表明,巨噬细胞中JNK的活化是肥胖引起胰岛素抵抗和炎症的必要条件[24]。AP-1转录因子也参与了LPS介导的巨噬细胞活化等炎症过程,同时AP-1的激活可与NF-κB协同作用,促使巨噬细胞向M1型巨噬细胞极化,起到促炎作用[25](图4)。

图4 AP-1对巨噬细胞极化的影响及与TLRs的相关性Fig.4 Effect of AP-1 on macrophage polarization and correlation with TLRs
2.5PPAR-γ PPAR-γ是一种配体激活的转录因子,属于核激素受体超家族,PPAR-γ在巨噬细胞中表达丰富,是巨噬细胞脂质代谢的主要调节因子,可通过多种机制抑制促炎基因的表达[26]。
IL-13和IL-4可诱导PPAR-γ的表达;PPAR-γ可协同STAT6促进单核细胞向M2型巨噬细胞极化;在肥胖或炎症应激时,PPAR-γ可导致脂肪组织巨噬细胞向M2型巨噬细胞极化;当PPAR-γ基因敲除时,在M1型巨噬细胞分泌的炎性因子IL-1β和TNF-α作用下,炎症和胰岛素抵抗进一步增强[27-29]。PPAR-γ与TLRs下游信号分子 NF-κB密切相关,越来越多的证据表明,大黄酚等药物活性分子的抗炎作用是通过PPAR-γ依赖的NF-κB的失活实现的[28,29](图5)。
2.6CREB和KLF4 CREB是一种选择性结合cAMP反应元件(cAMP response element,CRE)的核蛋白,含碱性亮氨酸拉链结构,其被蛋白激酶A(PKA)磷酸化后而活化,它的存在能刺激基因转录,故又被称为转录增强因子。研究发现,CREB能够通过诱导抗炎细胞因子IL-10的产生,增强M2型巨噬细胞的极化;前列腺素E2(prostaglandin E2,PGE2)通过活化cAMP/PKA/CREB途径,阻断M1巨噬细胞的极化,促进了M2巨噬细胞极化,所以,在急性营养过剩的情况下,巨噬细胞的CREB抑制了促炎因子的产生,有助于维持胰岛素的敏感性[30](图5)。
KLF4是一种含锌指结构的转录因子,发现在IL-4诱导的巨噬细胞活化过程中,STAT6的活化可激活KLF4,KLF4能够驱动许多M2表型基因的表达,且能够协同CREB,共同发挥抗炎应答作用[30]。在单核细胞向巨噬细胞极化的过程中,KLF4能够使载脂蛋白E(apolipoprotein E,apoE)基因启动子活性增高并诱导其表达,在CREB存在下,此作用进一步增强;apoE在脂代谢过程中具有重要作用,apoE缺陷可导致血浆中致动脉粥样硬化脂蛋白清除受阻,故KLF4可能是一个潜在的治疗动脉粥样硬化的新靶标;另外,TLRs通路活化产生的LPS和TNF-α等促炎因子是诱导巨噬细胞中KLF4表达的关键因素之一,进而使KLF4诱导抗炎因子表达,以维持内环境的稳态[31](图5)。
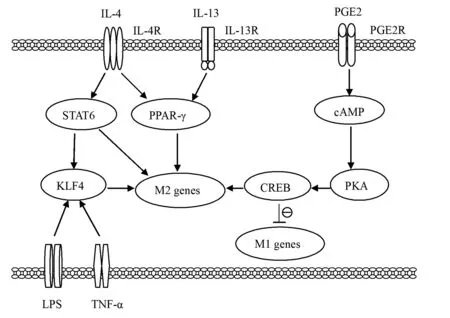
图5 PPAR-γ、CREB和KLF4对巨噬细胞极化的影响及与TLRs的相关性Fig.5 Effect of PPAR-γ,CREB and KLF4 on macrophage polarization and correlation with TLRs
综上,IFN-γ和LPS等促炎因子使巨噬细胞倾向M1表型,在转录因子STAT1、IRF1/IRF5/IRF8、p65/p50NF-κB和AP-1等作用下,促进促炎相关基因的表达,这些促炎因子使巨噬细胞更加倾向M1表型,形成促炎正反馈机制;同样,IL-4、IL-10和TGF-β等抗炎因子使巨噬细胞倾向M2表型,在转录因子STAT3/6、IRF3/IRF4/、p50/p50NF-κB、PPAR-γ、CREB和KLF4等作用下,诱导更多抗炎因子的表达,这些抗炎因子使巨噬细胞表型进一步向M2转移,形成抗炎正反馈机制;不同的转录因子间存在协同作用或反馈调控作用以调节极化方向;活化的TLRs信号通路可通过影响转录因子的活性,从而影响巨噬细胞极化方向。
上述这些转录因子介导的巨噬细胞极化参与了许多感染性疾病和炎症性疾病的发生、发展和转归。巨噬细胞极化机制复杂,TLRs存在嗜性差异,未来的研究应全面探索不同疾病介导巨噬细胞极化的转录因子表达谱,明确和转录因子交集的TLRs信号通路中的信号分子,以此挖掘相关疾病的治疗新靶点。
