利用WGCNA鉴定玉米株高和穗位高基因共表达模块
2020-02-20曹言勇王利锋李晶晶范艳萍李会勇
马 娟 曹言勇 王利锋 李晶晶 王 浩 范艳萍 李会勇
利用WGCNA鉴定玉米株高和穗位高基因共表达模块
马 娟 曹言勇 王利锋 李晶晶 王 浩 范艳萍 李会勇*
河南省农业科学院粮食作物研究所, 河南郑州 450002
株高和穗位高是玉米株型的重要影响因子, 与产量性状紧密相关。加权基因共表达网络分析(weighted gene co-expression network analysis, WGCNA)是探索基因网络与特定性状间关联关系的重要方法, 为株高和穗位高相关基因的挖掘提供新途径。本研究利用郑58、掖478、昌7-2和黄早四及其组配的杂交种郑单958、安玉5号、郑58/黄早四和掖478/黄早四, 结合其在45,000株 hm–2和67,500株 hm–2条件下的转录组数据, 采用WGCNA构建了2种密度条件下的共表达网络, 分别得到24个和21个共表达模块, 并鉴定到与株高和穗位高显著且高度相关(相关系数的绝对值>0.50)的共表达模块15个, 其中两性状相同的模块共6个。基因功能富集分析结果表明, 株高和穗位高共表达模块主要参与生长发育、光合作用、响应光刺激、植物激素、碳水化合物合成/代谢等重要活动。根据模块内基因的连接度, 发现乙烯响应因子、硫胺素酶、磷酸甘油酸激酶、谷胱甘肽转移酶和琥珀酸脱氢酶等是模块内的核心基因。通过构建其局部网络, 发现与已报道株高基因、和以及C3H转录因子、C2C2-GATA转录因子和乙烯受体同源子等存在关联关系。此外, 已报道株高基因和也存在于共表达模块中。以这5个已报道株高基因为核心, 构建其基因网路, 发现生长素转录因子、、、光合系统II氧进化多肽和光合系统I N亚基等与其存在关联。15个共表达模块和核心基因的挖掘以及基因生物学功能和互作网络的解析有助于揭示玉米株高和穗位高的遗传基础。
玉米; 加权基因共表达网络; 转录组; 株高; 穗位高
株高和穗位高是玉米株型的重要构成因子, 与产量密切相关。因此解析玉米株高和穗位高的遗传机制, 对玉米株型育种和产量的提高具有重要意义。前人利用数量性状定位和全基因组关联分析方法对玉米株高和穗位高的遗传构成开展了大量研究, 挖掘了许多与之相关的QTL/QTN[1-6], 也克隆了株高相关基因如()、、、、、()等[7-11]。尽管株高和穗位高研究取得了重要进展, 但从转录组水平解析其遗传构成的研究较少。
转录组研究是基因功能及结构研究的基础和出发点。加权基因共表达网络分析(weighted gene co-expression network analysis, WGCNA)是通过网络研究基因功能的重要方法[12]。将转录组数据与WGCNA方法结合可以挖掘出与特定性状相关的模块和基因。目前, 前人已经利用WGCNA方法研究了玉米籽粒大小和产量杂种优势等性状和组织特异性模块。Zhang等[13]发现13个与玉米粒重显著相关的模块, 其相关性为−0.87~0.89。Ma等[14]在67,500株 hm–2条件下挖掘出7个模块与玉米产量等性状杂种优势指数显著相关, 通过模块功能注释分析, 发现1461个基因可能与玉米杂种优势相关。Zhan等[15]发现胚乳隔间、糊粉层和胚乳等组织特异性模块10个。杨宇昕等[16]对B73不同发育阶段的转录组数据进行了WGCNA分析, 鉴定到14个组织特异性模块, 其中8个组织特异性模块内基因富集到与开花调控相关的代谢通路。此外, WGCNA也可用来挖掘非生物胁迫如镉胁迫[17]和淹水胁迫的相关基因[18]、氮响应的长链非编码RNA[19]和肌醇磷酸盐代谢涉及的新基因和调控机制[20]等。
本研究结合转录组测序(RNA-Seq)数据和WGCNA方法, 构建玉米加权基因共表达网络并划分模块, 挖掘株高和穗位高特异性模块, 通过对特异性模块分析, 挖掘株高和穗位高显著相关核心基因及其互作网络, 为进一步研究玉米株高和穗位高的分子机制提供新的思路。
1 材料与方法
1.1 材料和转录组测序数据
玉米自交系郑58、掖478、昌7-2、黄早四及其组配的杂交种郑单958 (郑58/昌7-2)、安玉5号(掖478/昌7-2)、郑58/黄早四和掖478/黄早四于2016年种植在河南省农业科学院新乡原阳试验田。采用随机区组试验设计, 3次重复。种植密度为45,000株 hm–2和67,500株 hm–2。授粉后15 d, 采集8个材料的穗位叶提取RNA, 用于转录组测序。采用Illumina X Ten对2种密度条件8个材料3个生物学重复进行双端测序。48个样品的转录组测序数据见NCBI (National Center for Biotechnology Information)的Sequence Read Archive数据库(登录号为 SRP136913)。利用Tophat v2.0.10将clean reads比对到玉米B73参考基因组(ftp://ftp.ensemblgenomes. org/pub/release-29/plants/fasta/zea_mays/dna/Zea_mays.AGPv3.29.dna.toplevel.fa.gz)。利用Cufflinks v.2.1.1计算基因在两种密度条件下8个材料3个生物学重复转录本的表达量。利用每千碱基外显子百万片段数(fragments per kilobase of exon per million fragments mapped, FPKM)值衡量基因的表达水平。在两种密度条件下, 分别选取24个样品FPKM值均大于1的基因, 用于构建基因共表达网络分析。授粉后45 d, 每个材料每个重复测量5个植株的株高和穗位高。利用R语言计算方差分析和Duncan’s多重比较。
1.2 加权基因共表达网络分析
利用R软件WGCNA包构建基因共表达网络分析(https://horvath.genetics.ucla.edu/html/Coexpression Network/Rpackages/WGCNA/index.html)。为了确保符合无尺度网络分布, WGCNA需要选择合适的加权系数β。β利用WGCNA包中的pickSoftThreshold函数计算。首先设置β=1–20, 分别计算其对应的相关系数和基因连接度均值。β的选取标准即满足相关系数的平方接近0.8, 同时还需要保证一定的基因连接度。根据图1结果, 在45,000株 hm–2和67,500株 hm–2条件下, β值均取9来构建共表达网络。采用动态切割法(dynamic tree cut)识别共表达模块。利用自动网络构建函数blockwiseModules构建网络, 其中最小模块大小(minModuleSize)为50, mergeCutHeight=0.25合并相似性为0.75的模块, 其他参数按照默认设置。利用softConnectivity函数计算基因的连接度, 筛选出模块内连接度前10的基因, 定义这些基因为模块的核心基因。根据拓扑重叠矩阵计算出模块内不同基因间的权重值。权重值越高表示基因间的关联程度越高。根据权重值, 选取与核心基因和已报道株高基因存在互作网络前10%的基因, 利用Cytoscape v3.6.1进行可视化展示。

图1 45,000株hm–2(A) 和67,500株 hm–2(B)条件下软阈值β的确定
A和B: 左图纵坐标是无尺度网络模型指数; 右图纵坐标每一个软阈值对应的平均连接度; 横坐标均代表软阈值β。
A and B: The ordinate represents the index of scale free network model in left figure. The ordinate represents the average link degree of each soft threshold in right figure. The abscissa represents the soft threshold β.
1.3 株高和穗位高显著关联模块的鉴定和基因功能分析
为了找到与株高和穗位高相关性较高的基因, 本研究定义相关系数绝对值在0.5以上且显著性达到0.05水平的模块为显著模块。利用R语言的clusterProfiler包对显著模块内基因进行GO (gene ontology)分析。以FDR=0.05为阈值来筛选显著生物学过程(biological process, BP)。
2 结果与分析
2.1 两种密度条件下8个材料株高和穗位高的表现
4个自交系和4个杂交种在67,500株 hm–2的株高和穗位高均高于45,000株 hm–2(图2)。只有掖478/黄早四的株高在两种密度条件下存在显著差异(双尾测验,=0.011)(图2-A)。方差分析结果显示同一密度条件下8个材料间存在显著差异(附表1和附表2)。Duncan’s多重比较结果表明同一密度条件下, 4个杂交种的株高和穗位高均显著高于4个自交系(图2)。两种密度条件下, 安玉5号的株高和穗位高均显著高于郑单958。2种密度条件下, 4个自交系的株高存在显著差异, 以昌7-2的株高最高、黄早四次之、掖478和郑58最低。两种密度条件下, 昌7-2和黄早四的穗位高没有显著差异, 掖478和郑58的穗位高也没有显著差异, 但昌7-2和黄早四的穗位高均显著高于掖478和郑58。

图2 45,000株 hm–2和67,500株hm–2条件下8个材料株高和穗位高
图中数字表示均值±标准误。柱中字母为Duncan’s多重比较结果, 不同字母表示材料间在< 0.05水平差异显著。*表示0.05显著水平。
Number represents mean±SE. Bars with different letters are significantly different at< 0.05 as determined by Duncan’s multiple comparison.*denotes significant at the 0.05 probability level.
2.2 基因共表达网络的构建
经过滤FPKM值<1的基因, 24个样品在45,000株 hm–2和67,500株 hm–2密度下, 分别有14,251个和14,190个基因用于构建加权基因共表达网络。首先根据基因的表达量进行聚类分析, 聚类程度高的基因将被分到一个模块。然后利用动态切割法识别共表达模块。不同的模块用不同的颜色表示。其中, Grey 模块表示未分配到任何模块的基因。低密度条件下共获得24个共表达模块(图3-A), 不同模块间包含的基因个数差异较大。其中, Turquoise模块的基因个数最多, 为3777, Darkturquoise模块的基因个数最少, 为57个(图4)。高密度条件下共获得21个模块(图3-B)。其中, Turquoise模块的基因个数最多(3283), Royalblue基因个数最少(71)(图5)。
2.3 株高和穗位显著相关的共表达模块及其功能注释
在45,000株hm–2条件下, 24个模块中与株高和穗位高相关性绝对值大于0.50, 且显著的模块分别有7个和4个, 两性状共同的模块有2个(图4)。其中, Turquoise模块与株高和穗位高均具有较高的相关性, 相关系数分别为−0.62 (=0.0013)和−0.80 (=2.33E-06)。Lightgreen模块与株高和穗位高均表现出显著负相关, 相关系数介于−0.51~ −0.64。Turquoise模块富集到较多生物学过程, 共有1667个BP条目(附图1)。其中215个条目与发育相关, 121个条目与转运相关, 67个条目与定位相关。Turquoise模块内基因还显著富集在信号途径(58)、信号传导(29)、碳水化合物代谢合成(44)、泛素相关的过程(23)、光合作用和光响应(24)以及激素相关的生物学过程(22)等。Lightgreen模块显著富集到255个BP条目, 其中43.92%为发育相关的过程(附图2)。

图3 45,000株hm–2(A)和67,500株hm–2(B)条件下基因聚类树和模块构建

图4 45,000株hm–2条件下株高(PH)和穗位高(EH)共表达模块及其基因个数和相关系数
在67,500株hm–2条件下, 与株高和穗位显著相关的共表达模块分别有4个和6个(图5)。其中, 与两性状均相关的基因模块有4个。Midnightblue和Pink模块与株高和穗位高均存在高度相关性, 相关系数介于−0.70~ −0.80。Purple和Tan模块与两性状也均有较高的相关性(= −0.64 ~ −0.72)。Midnightblue模块显著富集到362个BP条目, 主要包括发育相关过程(80)、碳水化合物代谢合成(18)、光合作用相关和光响应(15)、信号传导(15)、泛素相关的途径(11)和植物激素(3)等(附图3)。Pink模块基因也主要显著富集在发育相关(29)、定位(41)、光合作用(6)和光响应(3)等的生物学过程(附图4)。Purple模块基因主要富集在生物胁迫相关的过程(附图5)。Tan模块显著富集40个BP条目, 其中对光、热等刺激的响应有19条, 发育相关的条目有11条, 光合作用有2条(附图6)。

图5 67,500株hm–2条件下株高(PH)和穗位高(EH)共表达模块及其基因个数和相关系数
2.4 显著共表达模块的核心基因和已报道株高基因的基因网络
选取显著模块内连接度最高的前10个基因作为该模块的核心基因, 并对核心基因的互作网络利用Cytoscape软件进行了可视化。低密度条件下, Turquoise模块内连接度最高的前10个基因中, 有注释信息的基因为和硫胺素酶。是乙烯响应的转录因子, 可能是株高和穗位高重要的候选基因。由于与互作的基因较多, 共有2750个, 图6仅对权重值前150个有关联的基因进行可视化。与具有较高互作网络关系的基因为C3H转录因子、谷胱甘肽转移酶、已报道株高基因[21]、乙烯受体同源子和C2C2-GATA转录因子等。已知株高基因[10]和[22]与也存在关联, 但其权重值不在前10%中。的关联基因有、Alfin类转录因子、、肌动蛋白相关蛋白、F-box/kelch 重复蛋白SKIP11、FAR1相关序列、顺乌头酸、二硫键异构酶、核结合蛋白和亚油酸酯13S脂氧合酶等(图6)。在Lightgreen模块中, 核心基因有注释信息的是磷酸甘油酸激酶, 其关联的基因有SAR同源子和蛋白质二硫键异构酶等。67,500株 hm–2条件下, Pink模块核心基因是和琥珀酸脱氢酶。图6显示这2个基因间存在关联。与这2个基因均有网络关系的基因为、磷酸烯醇丙酮酸羧化酶、铝诱导蛋白同源子和叶绿体外膜的转位子等。
已报道株高相关基因如、和在45,000株hm–2条件下的Turquoise模块中。以这3个基因为核心构建局部网络, 发现Turquoise模块的核心基因与、和均存在互作(图7)。与、、半胱氨酸蛋白酶、半胱氨酸蛋白酶抑制剂、mTERF蛋白结构域和等存在关联。与、氯化钠胁迫蛋白和等表现出互作网络关系。与丙酮酸脱羧化酶和MAP激酶等存在互作关系。已报道株高基因[23]和[24]分别在Brown和Pink模块中。生长素转录因子和与关联。与光合系统II氧进化多肽、光合系统I N亚基、苯丙氨酸氨裂解酶和等存在互作网络关系。
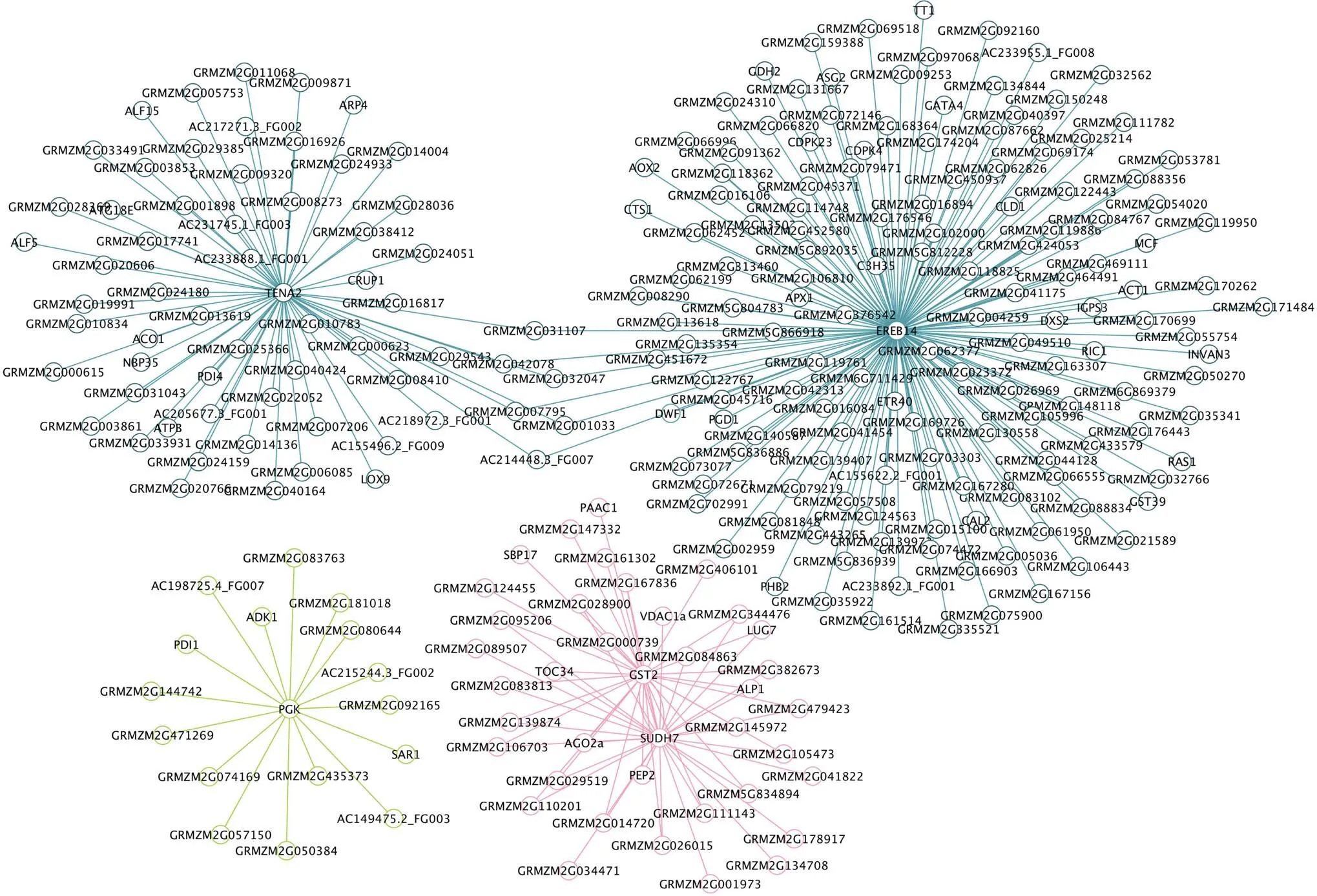
图6 显著共表达模块内核心基因的基因网络
图中青绿色、浅绿色和粉红色分别表示Turquoise、Lightgreen和Pink模块。
Colors in figure represent Turquoise, Lightgreen, and Pink modules.
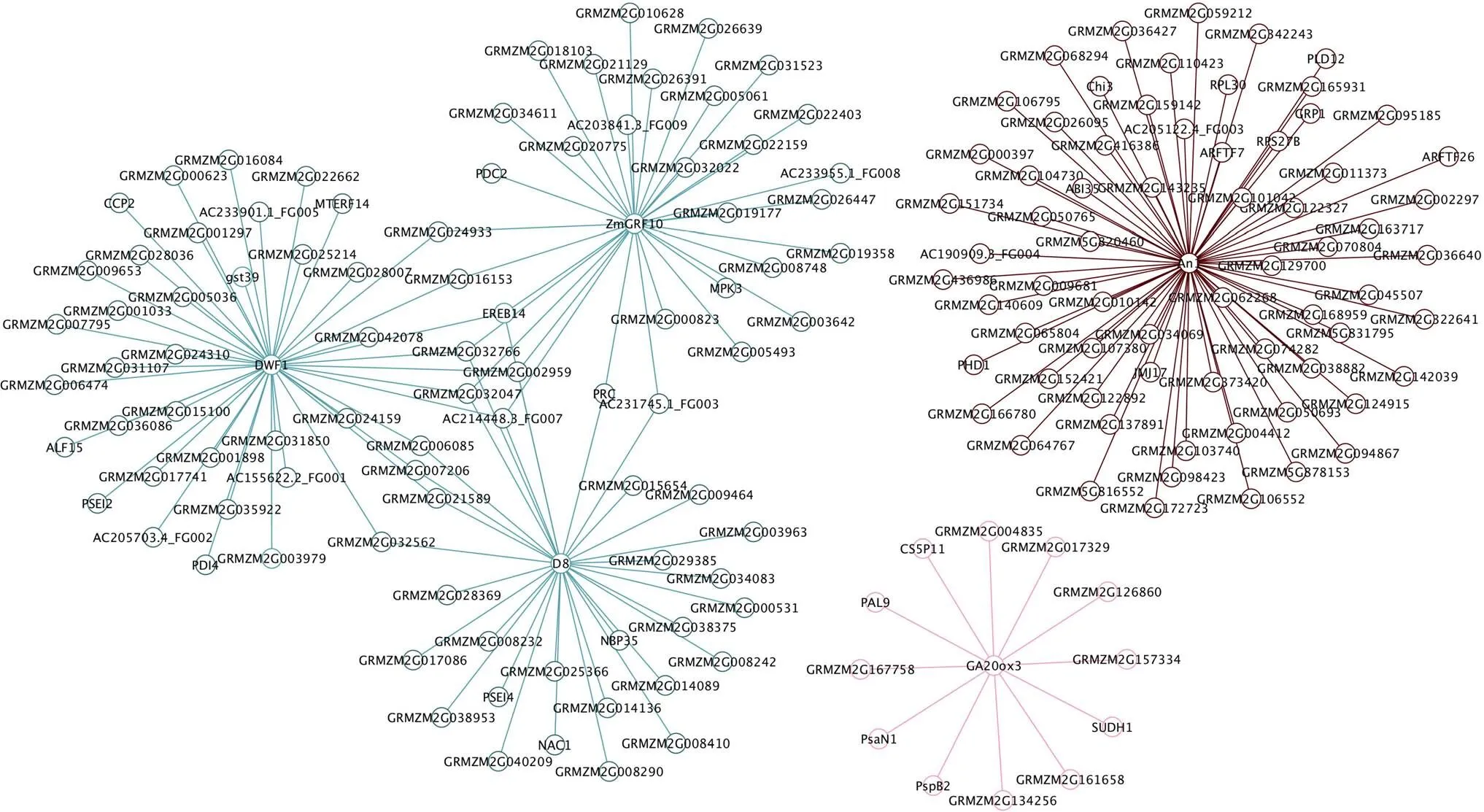
图7 已报道株高基因D8、DWF1、ZmGRF10、An1和GA20ox3的基因网络
图中青绿色、棕色和粉红色分别表示Turquoise、Brown和Pink模块。
Colors in figure represent Turquoise, Brown, and Pink modules.
3 讨论
随着测序技术的发展, 利用共表达网络对基因组、转录组和代谢组等海量组学数据进行系统研究, 是目前比较流行的大数据处理方法。WGCNA是共表达网络分析有效的分析方法, 能够特异地筛选出与目标性状具有高度生物学意义的共表达模块, 在玉米等植物中已经被证明是一种高效的数据挖掘方法[25]。
本研究利用WGCNA方法对8个玉米材料转录组测序数据构建了加权基因共表达网络, 两种密度条件下挖掘出株高和穗位高高度相关的模块共15个, 其中株高和穗位高相同的模块6个。通过对模块内基因的注释分析, 发现共表达基因主要与发育、光合作用、响应光刺激、植物激素和碳水化合物合成/代谢等重要活动相关。Wang等[26]对3个杂交种在拔节期、大喇叭口期和抽雄期的茎进行了转录组测序分析, 利用WGCNA挖掘了株高相关的共表达模块, 发现模块内基因主要参与植物激素相关代谢途径。已经克隆的株高基因如、和参与赤霉素合成和信号转导途径[8,10],基因参与调控生长素极性运输[11]。Wang等[26]挖掘的株高基因与本研究高低密度株高基因相同的分别有364个和1833个, 与高低密度穗位高基因相同的分别有725个和1722个。除了植物激素相关的途径外, Wang等也发现抽雄期3个杂交种间的差异基因显著富集在糖代谢和分生组织发育相关过程[26]。因此, 这些重要的生物学活动可能与株高和穗位高的形成有关。
根据基因的连接度, 我们筛选了株高和穗位高显著模块内的核心基因, 并对其基因网络进行了研究。其中, 乙烯响应因子是低密度条件下株高和穗位高显著相关模块Turquoise的核心基因。乙烯响应因子家族在激素、非生物学胁迫和生长发育等方面具有重要作用[27-29]。本研究中也显著富集在生长素响应和其刺激响应、生长素介导的信号途径、发育过程、非生物学胁迫和碳水化合物代谢等过程。该基因与已报道株高基因具有较高的关联关系。其他株高基因和也与存在关联。这些表明可能是影响株高的重要基因, 但其作用机制有待进一步探究。
已报道的株高相关基因如、、、和均存在于本研究株高和穗位高共表达模块中。其中、和在45,000株hm–2条件下株高和穗位高显著相关的Turquoise模块中。是赤霉素响应突变体基因, 其突变体在DELLA区、VHYNP区及同时这2个区发生氨基酸不等数目的缺失导致赤霉素信号传导受阻而产生矮秆或半矮秆表型[10,30]。是拟南芥油菜素类固醇合成基因的同源基因, 其转基因玉米植株表现出不同程度的矮秆情况[21]。是生长调节转录因子, 其过表达会通过降低细胞增殖从而导致玉米植株的降低和叶片的变小, 但产量相关性状并没有发生改变[22]。除了前面提到的Turquoise模块的核心基因外, 我们发现很多转录因子与这3个株高基因存在网络关系。例如C2C2-CO-like转录因子与这3个株高基因均存在关联关系。bZIP转录因子、、、和与和均存在互作关系。和与和存在互作网络关系。同源框转录因子和分别与和具有关联关系。尽管这些转录因子与已报道株高基因的权重值不在其局部网络的前10%内, 但其与已知株高基因网络关系的发掘有助于揭示株高的遗传机制。
和也是株高相关的基因, 分别在Brown和Pink模块中。通过影响油菜素类固醇合成而导致玉米茎节间长度缩短[23]。生长素转录因子和与具有互作关系。在拟南芥中,受到远红外光的刺激会导致叶柄伸长[24], 进而导致植株高度变化。已克隆株高基因是调控玉米株高的主效QTL的候选基因[31]。赤霉素累积会引起该主效QTL染色体片段代换系的节间增长[31]。Pink模块中,与光合系统II氧进化多肽、光合系统I N亚基、苯丙氨酸氨裂解酶和等存在互作关系。
此外, QTL定位和全基因组关联分析方法鉴定的株高和穗位高候选基因也在本研究的共表达模块内。例如, 郑雷等在元分析整合的株高一致性QTL置信区间内发掘的候选基因(编码棕色叶中脉)[32]存在于45,000株hm–2下显著模块Lightyellow中。45,000株hm–2下发现的与株高显著相关的基因是转录因子, 也是穗位高显著关联的SNP (SYN32392)的候选基因[4]。45,000株hm–2下发现的株高显著相关基因(线粒体底物载体家族蛋白)和(亮氨酸重复蛋白激酶家族蛋白)分别是株高显著关联SNP 位点S3_18231963和S6_ 164148781的候选基因[5]。而且, 这2个SNP解释株高表型变异率比较高, 均为9.3%[5]。45,000株hm–2条件下的株高和穗位高共同基因是穗位高显著关联位点S2_25387853的候选基因[5]。67,500株hm–2条件下的株高和穗位高共同基因(转导蛋白/WD40重复超家族蛋白)是Li等[5]研究中穗位高关联位点S5_69308290的候选基因。
上述前人研究结果与本研究株高和穗位高共表达模块基因存在诸多重合, 说明利用WGCNA从转录组水平鉴定株高和穗位高共表达模块及其生物学意义是可行的。因此, 利用WGCNA方法解析株高和穗位高等重要农艺性状, 分析其共表达模块的生物学功能, 挖掘显著关联模块内的关键基因, 可以为解析复杂农艺性状的分子机制提供重要途径。
4 结论
利用WGCNA方法构建了不同密度条件下株高和穗位高的加权共表达网络, 得到高度关联模块15个。揭示了株高和穗位高关联模块的生物学功能, 鉴定了关联模块内核心基因, 解析了其基因网络关系。
[1] 李清超, 李永祥, 杨钊钊, 刘成, 刘志斋, 李春辉, 彭勃, 张岩, 王迪, 谭巍巍, 孙宝成, 石云素, 宋燕春, 张志明, 潘光堂, 黎裕, 王天宇. 基于多重相关RIL群体的玉米株高和穗位高QTL定位. 作物学报, 2013, 39: 1521–1529. Li Q C, Li Y X, Yang Z Z, Liu C, Liu Z Z, Li C H, Peng B, Zhang Y, Wang D, Tan W W, Sun B C, Shi S Y, Song C Y, Zhang Z M, Pan G T, Li Y, Wang T Y. QTL mapping for plant height and ear height by using multiple related RIL populations in maize., 2013, 39: 1521–1529 (in Chinese with English abstract).
[2] 何坤辉, 常立国, 崔婷婷, 渠建洲, 郭东伟, 徐淑兔, 张兴华, 张仁和, 薛吉全, 刘建超. 多环境下玉米株高和穗位高的QTL定位. 中国农业科学, 2016, 49: 1443–1452. He K H, Chang L G, Cui T T, Qu J Z, Guo D W, Xu S T, Zhang X H, Zhang R H, Xue J Q, Liu J C. Mapping QTL for plant height and ear height in maize under multi-environments., 2016, 49: 1443–1452 (in Chinese with English abstract).
[3] 刘坤, 张雪海, 孙高阳, 闫鹏帅, 郭海平, 陈思远, 薛亚东, 郭战勇, 谢惠玲, 汤继华, 李卫华. 玉米株型相关性状的全基因组关联分析. 中国农业科学, 2018, 51: 821–834. Liu K, Zhang X H, Sun G Y, Yan P S, Guo H P, Chen S Y, Xue Y D, Guo Z Y, Xie H L, Tang J H, Li W H. Genome-wide association studies of plant type traits in maize., 2018, 51: 821–834 (in Chinese with English abstract).
[4] 李凯, 张晓祥, 管中荣, 沈亚欧, 潘光堂. 玉米株高和穗位高的全基因组关联分析. 玉米科学, 2017, 25(6): 1–7. Li K, Zhang X X, Guan Z R, Shen Y O, Pan G T. Genome-wide association analysis of plant height and ear height in maize., 2017, 25(6): 1–7 (in Chinese with English abstract).
[5] Li X, Zhou Z, Ding J, Wu Y, Zhou B, Wang R, Ma J, Wang S, Zhang X, Xia Z, Chen J, Wu J. Combined linkage and association mapping reveals QTL and candidate genes for plant and ear height in maize., 2016, 7: 833.
[6] Weng J, Xie C, Hao Z, Wang J, Liu C, Li M, Zhang D, Bai L, Zhang S, Li X. Genome-wide association study identifies candidate genes that affect plant height in Chinese elite maize (L.) inbred lines., 2011, 6: e29229.
[7] Fujioka S, Yamane H, Spray C R, Gaskin P, Macmillan J, Phinney B O, Takahashi N. Qualitative and quantitative analyses of gibberellins in vegetative shoots of normal,,,, andseedlings ofL., 1988, 88: 1367–1372.
[8] Winkler R G, Helentjaris T. The maizegene encodes acytochrome P450-mediated early step in gibberellin biosynthesis., 1995, 7: 1307–1317.
[9] Thornsberry J M, Goodman M M, Doebley J, Kresovich S, Nielsen D, Buckler E S.polymorphisms associate with variation in flowering time., 2001, 28: 286–289.
[10] Lawit S J, Wych H M, Xu D, Kundu S, Tomes D T. Maize DELLA proteinsandas modulators of plant development., 2010, 51: 1854–1868.
[11] Multani D S, Briggs S P, Chamberlin M A, Blakeslee J J, Murphy A S, Johal G S. Loss of an MDR transporter in compact stalks of maizeand sorghummutants., 2003, 302: 81–84.
[12] Zhang B, Horvath S. A general framework for weighted gene co-expression network analysis., 2005, 4: Article 17.
[13] Zhang X, Hirsch C N, Sekhon R S, De Leon N, Kaeppler S M. Evidence for maternal control of seed size in maize from phenotypic and transcriptional analysis., 2016, 67: 1907–1917.
[14] Ma J, Zhang D, Cao Y, Wang L, Li J, Lübberstedt T, Wang T, Li Y, Li H. Heterosis-related genes under different planting densities in maize (L.)., 2018, 69: 5077–5087.
[15] Zhan J, Thakare D, Ma C, Lloyd A, Nixon N M, Arakaki A M, Burnett W J, Logan K O, Wang D, Wang X, Drews G N, Yadegari R. RNA sequencing of laser-capture microdissected compartments of the maize kernel identifies regulatory modules associated with endosperm cell differentiation., 2015, 27: 513–531.
[16] 杨宇昕, 桑志勤, 许诚, 代文双, 邹枨. 利用WGCNA进行玉米花期基因共表达模块鉴定. 作物学报, 2019, 45: 161–174. Yang Y X, Sang Z Q, Xu C, Dai W S, Zou C. Identification of maize flowering gene co-expression modules by WGCNA., 2019, 45: 161–174 (in Chinese with English abstract).
[17] Peng H, He X, Gao J, Ma H, Zhang Z, Shen Y, Pan G, Lin H. Transcriptomic changes during maize roots development responsive to Cadmium (Cd) pollution using comparative RNA seq- based approach., 2015, 464: 1040–1047.
[18] Thirunavukkarasu N, Hossain F, Mohan S, Shiriga K, Mittal S, Sharma R, Singh R K, Gupta H S. Genome-wide expression of transcriptomes and their co-expression pattern in subtropical maize (L.) under waterlogging stress., 2013, 8: e70433.
[19] Lyu Y, Liang Z, Ge M, Qi W, Zhang T, Lin F, Peng Z, Zhao H. Genome-wide identification and functional prediction of nitrogen-responsive intergenic and intronic long non-coding RNAs in maize (L.)., 2016, 17: 350.
[20] Zhang S, Yang W, Zhao Q, Zhou X, Jiang L, Ma S, Liu X, Li Ye, Zhang C, Fan Y, Chen R. Analysis of weighted co-regulatory networks in maize provides insights into new genes and regulatory mechanisms related to inositol phosphate metabolism., 2016, 17: 129–146.
[21] Tao Y, Zheng J, Xu Z, Zhang X, Zhang K, Wang G. Functional analysis of, a maize homolog of thebrassinosteroids biosynthetic DWF1/DIM gene., 2004, 167: 741–751.
[22] Wu L, Zhang D, Xue M, Qian J, He Y, Wang S. Overexpression of the maize, an endogenous truncated growth regulating factor protein, leads to reduction in leaf size and plant height., 2014, 56: 1053–1063.
[23] Hartwig T, Chuck G S, Fujioka S, Klempien A, Weizbauer R, Potluri D P, Choe S, Johal G S, Schulz B. Brassinosteroid control of sex determination in maize., 2011, 108: 19814–19819.
[24] Tamotsu H, Rod W K, Chris A H, Masaji K. The involvement of gibberellin 20-oxidase genes in phytochrome-regulated petiole elongation of., 2005, 138: 1106–1116.
[25] Zhao W, Langfelder P, Fuller T, Dong J, Li A, Hovarth S. Weighted gene coexpression network analysis: state of the art., 2010, 20: 281–300.
[26] Wang H, Gu L, Zhang X, Liu M, Jiang H, Cai R, Zhao Y, Cheng B. Global transcriptome and weighted gene co-expression network analyses reveal hybrid-specific modules and candidate genes related to plant height development in maize., 2018, 98: 187–203.
[27] Mizoi J, Shinozaki K, Yamaguchi-Shinozaki K. AP2/ERF family transcription factors in plant abiotic stress responses., 2012, 1819: 86–96.
[28] Hinz M, Wilson I W, Yang J, Buerstenbinder K, Llewellyn D, Dennis E S, Sauter M, Dolferus R.RAP2: 2. An ethylene response transcription factor that is important for hypoxia survival., 2010, 153: 757–772.
[29] Licausi F, Ohme Takagi M, Perata P. APETALA2/ethylene responsive factor (AP2/ERF) transcription factors: mediators of stress responses and developmental programs., 2013, 199: 639–649.
[30] Cassani E, Bertolini E, Cerino B F, Landoni M, Gavina D, Sirizzotti A, Pilu R. Characterization of the first dominant dwarf maize mutant carrying a single amino acid insertion in the VHYNP domain of thegene., 2009, 24: 375–385.
[31] Teng F, Zhai L, Liu R, Bai W, Wang L, Huo D, Tao Y, Zheng Y, Zhang Z., a candidate gene for a major QTL,, for plant height in maize., 2013, 73: 405–416.
[32] 郑雷, 周羽, 曾兴, 邸宏, 翁建峰, 李新海, 王振华. 玉米株高QTL定位研究进展. 作物杂志, 2016, (2): 8–13.Zheng L, Zhou Y, Zeng X, Di H, Weng J F, Li X H, Wang Z H. QTL Mapping of plant height in maize., 2016, (2): 8–13 (in Chinese with English abstract).
Identification of gene co-expression modules of maize plant height and ear height by WGCNA
MA Juan, CAO Yan-Yong, WANG Li-Feng, LI Jing-Jing, WANG Hao, FAN Yan-Ping, and LI Hui-Yong*
Institute of Cereal Crops, Henan Academy of Agricultural Sciences, Zhengzhou 450002, Henan, China
Plant height (PH) and ear height (EH) are important factors for maize plant type and grain yield. Weighted gene co-expression network analysis (WGCNA) is an important method to explain the relationships between gene network and complicated traits and identify the PH and EH associated genes. In this study, we used Zheng 58, Ye 478, Chang 7-2, Huangzaosi and its combinations Zhengdan 958, Anyu 5, Zheng 58/Huangzaosi, and Ye 478/Huangzaosi as materials and utilized transcriptome data under the planting densities of 45,000 plants hm–2and 67,500 plants hm–2to construct a co-expression network by WGCNA, getting 24 and 21 co-expression modules, respectively. Among them, a total of 15 co-expression modules were significantly correlated with PH and EH, with the absolute correlation coefficients higher than 0.50. Six modules were overlapped between PH and EH. By gene function analysis, these overlapped modules were significantly enriched in development, photosynthesis, response to light stimulus, plant hormone, and carbohydrate biosynthesis/metabolism related activities. According to connectivity of genes in modules, AP2-EREBP transcription factor, thiaminase, phosphoglyceric kinase, glutathione transferase, and succinate dehydrogenasewere considered as hub genes. From gene networks,was connected with three known PH genes,,, and(C3H transcription factor),(C2C2-GATA transcription factor), and ethylene homology. Reported PH genesandwere also found in our co-expression modules. From the networks of the five known PH genes, ARF-transcription factor 7 (),,, photosystem II oxygen evolving polypeptide, and photosystem I N subunithad connections with these known PH genes. The identification of 15 co-expression modules and their hub genes, and analysis of their gene function and gene networks of key genes will be helpful for revealing the genetic basis of PH and EH.
maize; weighted gene co-expression network; transcriptome; plant height; ear height
2019-04-02;
2019-09-26;
2019-10-10.
10.3724/SP.J.1006.2020.93021
李会勇, E-mail: lihuiyong1977@126.com
E-mail: majuanjuan85@126.com
本研究由国家重点研发计划项目(2016YFD100103)和河南省科技攻关项目(192102110008)资助。
This study was supported by the National Key Research and Development Program of China (2016YFD100103) and the Science and Technology Project of Henan province (192102110008).
URL:http://kns.cnki.net/kcms/detail/11.1809.S.20191010.1139.002.html
