利用EST-SSR标记评价羽扇豆属(Lupinus L.)遗传多样性
2020-02-20张红岩张力科于海天胡锦国峰王何玉华宗绪晓
张红岩 杨 涛 刘 荣 晋 芳 张力科 于海天胡锦国 杨 峰王 栋 何玉华,* 宗绪晓,*
利用EST-SSR标记评价羽扇豆属(L.)遗传多样性
张红岩1,5,**杨 涛1,**刘 荣1,**晋 芳2张力科2于海天3胡锦国4杨 峰3王 栋1何玉华3,*宗绪晓1,*
1中国农业科学院作物科学研究所/ 国家农作物基因资源与基因改良重大科学工程, 北京 100081;2全国农业技术推广服务中心, 北京 100125;3云南省农业科学院粮食作物研究所, 云南昆明 650205;4USDA-ARS Western Regional Plant Introduction Station (WRPIS), Pullman, WA 99164, USA;5青海大学, 青海西宁 810016
利用基于窄叶羽扇豆转录组开发并筛选出的95对多态性EST-SSR标记, 对羽扇豆属的22个种133份资源进行全基因组扫描, 初步探究羽扇豆属下种间的进化关系, 分析来源于“旧世界”羽扇豆种间的遗传多样性, 为羽扇豆优异资源的挖掘和创新利用提供理论依据。结果表明, 用95对SSR标记共检测出1318个等位变异, 每对标记平均检测出3~37个等位变异, 平均为13.87个; 多态性信息量(PIC)变化范围0.39~0.91, 平均为0.75; 基因多样性变化范围0.41~0.92, 平均为0.78。基于邻接法(NJ)的系统发育初步探究了羽扇豆种间的进化关系, 22个羽扇豆种分别来源于“旧世界”和“新世界”, 与之前研究结果相对一致。聚类分析、群体结构分析和主成分分析结果均表明, 来源于“旧世界”的7个羽扇豆种被划分为4个组群, 各组群所包含的参试资源没有种间交叉重叠。
EST-SSR; 羽扇豆属; 转录组; 遗传多样性
羽扇豆(lupin或lupine), 俗称“鲁冰花”, 是一种富含蛋白, 能够固定大气游离氮、有效吸收土壤磷, 且具重要观赏价值的豆科植物。羽扇豆拥有悠久的栽培历史, 但尚未在中国发现原始栽培种。羽扇豆属是一个相对较大的属, 世界上约有300个种[1], 包括一年生和多年生种, 广泛分布于世界各地, 大部分来自“新世界”(New World, 简称NW), 只有13个种(L.、L.、var.Boiss. & Sprun.、L.、L. subsp.Desv.、Gladst.、Guss.、Forsk.、Boiss. & Reuter.、L.、Guss.、Boiss.、Murr.)来自欧洲、北非和东非[2]。遗传相似性研究表明, 羽扇豆多样性中心可分为: (1)美洲北部和中部, 安第斯美洲南部; (2)美洲南部(大西洋); (3)地中海沿岸地区及北非和东非地区[3]。由于喹啉类生物碱和抗营养化合物的存在, 大多数野生羽扇豆都具有苦味和毒性。目前, 栽培种羽扇豆均为一年生, 包括来源于“旧世界” (Old World, 简称OW)的窄叶羽扇豆(2= 40,L.)、白羽扇豆(2= 50,Forsk.)、沙质平原羽扇豆(2= 32,Guss.)、黄羽扇豆(2= 52,L.)等羽扇豆以及来源于“新世界”的南美羽扇豆(2= 48,Sweet.)。
在以往的研究中, 大多利用形态和农艺性状[4]、同工酶[5]、核糖体RNA ITS1和ITS2[6-7]、叶绿体基因[8]以及AFLP、ISSR、RAPD、DART和SSR[9-11]分析羽扇豆属(genus)的系统发育和遗传多样性。Drummond等[12]利用5个核基因、2个叶绿体基因和4个叶绿体非编码区完成了羽扇豆属的系统发育和遗传多样性分析。Nevado等[13]利用RNA-seq技术分析了来源于新世界的55个羽扇豆种中的67份材料, 结果表明, 蛋白质序列和基因表达水平的不同是造成羽扇豆属具有丰富遗传多样性的重要原因。
SSR (simple sequence repeats)是一种以特异引物PCR为基础的分子标记, 具有共显性、多态性高、分布广泛、重复性好等优点, 目前已广泛应用于主要农作物的分子遗传学研究, 然而尚未见规模性开发EST-SSR引物并用于羽扇豆属下种间遗传多样性和亲缘关系分析的报道。本研究利用基于窄叶羽扇豆(L.)转录组开发筛选得到的95对多态性EST-SSR标记, 初步分析了羽扇豆属(genus)的系统发育; 深入研究了来源于“旧世界”羽扇豆属内主要栽培种以及其他类型种质资源的群体结构、遗传变异及遗传进化关系, 剖析了种间的遗传关系, 以期为今后羽扇豆种质资源的搜集与利用、主要种的系统学研究以及挖掘优良品种等提供理论依据。
1 材料与方法
1.1 植物材料
供试羽扇豆材料133份, 22个种, 分布于六大洲, 其中7个种(77份材料)来源于“旧世界”。参试材料均来源于美国西部地区植物引种站(Western Regional Plant Introduction Station, Pullman, WA)(附表1), 2017年种植于云南省农业科学院试验场(25°12′N, 102°77′E), 常规大田管理。
利用DIVA-GIS[14](http://diva-gis.org/)绘制供试材料原始来源地(图1)。
1.2 DNA提取
大田播种后40 d, 选择每份参试材料生长良好的5个单株, 取幼嫩叶片混合, 利用改良CTAB法[15-16]提取DNA, 提取缓冲液中加入一定量的水溶性PVP和β-羟基乙醇, 可以有效防止酚类物质的氧化作用。用1.0%的琼脂糖凝胶电泳检测其质量, NanoDrop 2000检测DNA浓度, 根据检测结果将其稀释到工作液浓度50 ng μL-1后于-20℃保存备用。
1.3 SSR开发及多态性标记筛选
基于窄叶羽扇豆转录组数据(http://www.lupin- express.org/node/16), 利用SSR Locator software[17]检测SSR位点, 随机设计800对EST-SSR引物。SSR引物设计参数为, 两核苷酸至少重复4次, 三核苷酸至少重复3次, 引物长度18~22 bp, GC含量40%~55%, 引物退火温度55~65℃, 扩增片段长度100~400 bp。
从22个羽扇豆种中随机各选一份材料(附表1), 利用8%非变性聚丙烯酰胺凝胶电泳共筛选出95对多态性SSR标记(附表2)。委托北京梓熙生物科技有限公司合成SSR引物及荧光检测PCR扩增产物。
1.4 PCR扩增反应及产物检测
扩增反应总体积为10 μL, 包含1.5 μL基因组DNA (50 ng μL-1)、5 μL 2×PCR Master Mix (Genstar, 北京, 中国)、0.5 μL正向引物(2 μmol L-1)[5¢端标记荧光素FAM (蓝色)或Hex (绿色)]、0.5 μL反向引物(2 μmol L-1)和2.5 μL ddH2O。PCR 40 s, 程序为95℃预变性5 min; 95℃变性35 s, 54℃退火72℃延伸45 s, 34个循环; 72℃延伸5 min, 10℃保温7 min。将FAM (蓝色)和HEX (绿色)荧光标记的PCR扩增产物用超纯水稀释10~30倍, 分别取等体积的上述2种稀释液混合形成混合液, 吸取1 μL混合液, 分别加0.1 μL LIZ500分子量内标和8.9 μL去离子甲酰胺于DNA分析仪专用深孔板孔中; 然后将其在PCR仪上95℃变性5 min, 取出后立即置碎冰上, 冷却15 min左右; 瞬时离心10 s, 在ABI3700 DNA遗传分析仪上检测。

图1 基于不同地理来源的羽扇豆资源分布图
1.5 数据分析
1.5.1 数据收集和标记定位 利用DNA Collection和Gene Mapper软件收集和分析原始数据, 建立相应数值数据库。在统计和收集数据时, 若在某个种的所有材料中均不存在某个位点的任何等位基因,则表明该羽扇豆种不存在相应的SSR位点, 则记录为“9999”。通过BLAST比对获得95对多态性EST-SSR标记在窄叶羽扇豆(L.)染色体上的详细位置(https://www.ncbi.nlm.nih.gov/ genome/11024)。
利用MapInspect (http://mapinspect.software. informer.com/)绘制标记在染色体上的位置(图2)。
1.5.2 标记多态性及羽扇豆系统发育分析 采用PowerMarker 3.25软件[18]计算基于133份羽扇豆材料(附表1)的各引物对的等位基因数(No. of allele)、基因多样性(gene diversity)和多态性信息量(polymorphism information content, 简称PIC)(表1)。从133份羽扇豆材料中随机选取93份资源(附表1)用于初步分析羽扇豆属系统发育, 若某个种内材料份数很少, 则分析该种的全部材料; 若某个种内材料份数较多, 则随机选取几份。基于22个羽扇豆种的93份材料的等位基因频率和遗传距离, 利用邻接法(Neighbor-Joining)构建羽扇豆属的系统发育树(bootstrap设为1000), 用MEGA v5[19]绘制树图。
1.5.3 77份羽扇豆资源的遗传多样性分析 羽扇豆栽培种主要来源于“旧世界”, 为了探究羽扇豆栽培种的群体结构、遗传变异及遗传进化关系, 本研究选取来源于“旧世界”的7个羽扇豆种(包括4个主要栽培种和其他3个非栽培种)的77份(附表1)资源, 剖析参试种间的遗传关系, 为今后主要栽培种的遗传多样性研究以及挖掘优良品种等提供理论依据。利用PopGen version 1.32软件[20]计算7个羽扇豆种的Nei’s遗传一致度和遗传距离, 采用MEGA v5, 基于UPGMA (Unweighted Pair Group Method with Arithmetic Means)绘制聚类图。用Power Marker v3.25计算77份资源间的Nei’s遗传距离, FigTree v1.4.3 (http://tree.bio.ed.ac.uk/software/figTree/)绘制基于邻接法(Neighbor-Joining)的树形图。利用Structure 2.2软件[21]进行群体遗传结构分析, Burnin Period和after Burnin各为10,000,值为1~10, 每个值各运行15次, 利用在线软件Structure Harvester确定最佳组群数()。通过GenALEX V6.5[22]进行羽扇豆种间分子方差分析(AMOVA)、遗传分化系数(st)和主成分分析(PCA)。

图2 95对EST-SSR标记在窄叶羽扇豆染色体上分布情况

表1 95多态性标记遗传参数

(续表1)
PIC表示多态性信息量。PIC means polymorphism information content.
2 结果与分析
2.1 SSR标记多态性分析
筛选出95对扩增稳定、多态性较好的EST-SSR标记, 它们分布在20条染色体上, 每条染色体平均4.75个标记(图2)。NLL-09标记数最多(9), 其次是NLL-01和NLL-12, 均有8个标记, NLL-20标记数最少(1)。133份羽扇豆资源在95个SSR位点共检测出1318个等位变异, 平均每对引物5~37个等位变异, 平均为13.87个。PIC指数在0.39~0.91之间, 平均值为0.75; 基因多样性在0.41~0.92之间, 平均值为0.78。其中, lup-est 3649获得的等位基因数目最多(37), PIC值为0.91, 基因多样性为0.90; 其次是lup-est 6567(31)和lup-est 2531(30), PIC值分别为0.91和0.89, 基因多样性分别为0.91和0.88; 等位基因数目最少的是lup-est 3576(5), PIC值为0.66, 基因多样性为0.71 (表1)。上述结果表明, 本研究所用的EST-SSR标记满足遗传多样性分析的基本要求。
2.2 羽扇豆属系统发育分析
系统发育(bootstrap = 1000)关系表明, 22个羽扇豆种大致分为两类, 一类来源于“旧世界”, 包括; 另一类来源于“新世界”(NW), 包括。除外, 其他种的分类结果与前人研究结论一致[2,12](图3)。
2.3 77份羽扇豆资源聚类分析
利用PopGen version 1.32软件计算来源于旧世界的7个羽扇豆种间的遗传距离和遗传一致度, MEGA v5绘制物种聚类图。
基于UPGMA聚类方法, 7个羽扇豆种在遗传距离值为0.720时被明显划分为4个组群(图4), 组群I ()含; 组群II ()含; 组群III ()含、和; 组群IV ()含和。由表2可知, 7个羽扇豆种间平均遗传距离为0.411~1.845, 遗传一致度为0.158~0.633。其中和间的遗传距离最大(1.845), 遗传一致度最低(0.158);和间的遗传距离最小(0.411), 遗传一致度最高(0.663)。
基于遗传一致度分析来源于旧世界的4个栽培种之间亲缘关系表明, 白羽扇豆()和黄羽扇豆()(0.263)>黄羽扇豆()和窄叶羽扇豆()(0.254)>窄叶羽扇豆()和白羽扇豆()(0.244)>白羽扇豆()和沙质平原羽扇豆()(0.199)>黄羽扇豆()和沙质平原羽扇豆()(0.167)>窄叶羽扇豆()和沙质平原羽扇豆()(0.158)。基于邻接法(Neighbor-Joining), 77份羽扇豆资源同样被明显地划分为4个组群(图5-d), 与基于种子类型划分的组群完全一致。
2.4 77份羽扇豆资源群体结构分析
为了探讨参试个体间的群体结构关系, 利用Structure软件分析77份羽扇豆种质资源的遗传结构, 因后验概率值(ln())随亚群数的增大而增大, 故采用基于D的最大似然估计确定最适组群数()(图5-a)。当某个材料的Q值大于0.75时, 将该材料划分到相应的组群, 此时该材料的血缘关系相对单一。77份参试材料在= 4时,D出现峰值(图5-b), 即参试材料可被划分为4个组群(图5-c)。进一步分析群体结构的各组群成分, 表明各组群所包含的羽扇豆种类与聚类结果完全一致。白羽扇豆、黄羽扇豆、窄叶羽扇豆、沙质平原羽扇豆分属4个不同的群组, 表明4个栽培种之间有明显的种间隔离。
2.5 77份羽扇豆资源主成分分析(PCA)
为了更好地反映羽扇豆资源间的遗传关系, 利用GenALEX软件对77份羽扇豆种质资源进行主成分分析, 根据第一、第二维主成分绘制出二维主成分结构图(图5-e), 前三位特征向量PC1 (38.93%)、PC2 (26.10%)和PC3 (17.48%)占遗传变异的82.51%。PC1~PC2二维主成分结构图显示, 77份材料被明显地划分为4个组群, 4个组群之间具有明显的分界, 没有重叠部分。这一结果与聚类分析和群体结构分析结果完全一致, 进一步说明了组群分类结论的可靠性。
2.6 77份羽扇豆资源分子方差分析(AMOVA)
为了全面、准确地评价7个羽扇豆种间与种内的遗传变异, 利用GenALEX软件对77份参试材料进行分子方差(AMOVA)和遗传分化(st)分析。7个羽扇豆种在可信度= 0.01条件下, 羽扇豆种间和种内的遗传变异分别占总变异的80.00%和20.00% (表3), 羽扇豆种间存在明显的遗传结构, 种间遗传变异是总遗传变异的主要来源。
羽扇豆种间的遗传分化系数(st)在0.320~ 0.743之间, 存在较大差异, 说明羽扇豆种群的遗传分化较大。由表4可知,和遗传分化程度最大(0.743);和遗传分化程度最小(0.320)。上述结果与基于遗传距离得到聚类结果(图4)一致性较高, 说明羽扇豆属的遗传变异主要由种间遗传差异引起。

图4 基于7个羽扇豆不同种间(“旧世界”)的UPGMA 聚类图

表2 7个羽扇豆种间(“旧世界”)的Nei’s (1978)遗传距离和遗传一致度
对角线以上表示遗传一致度; 对角线以下表示遗传距离。
The genetic identity is shown above diagonal and genetic distance below diagonal.
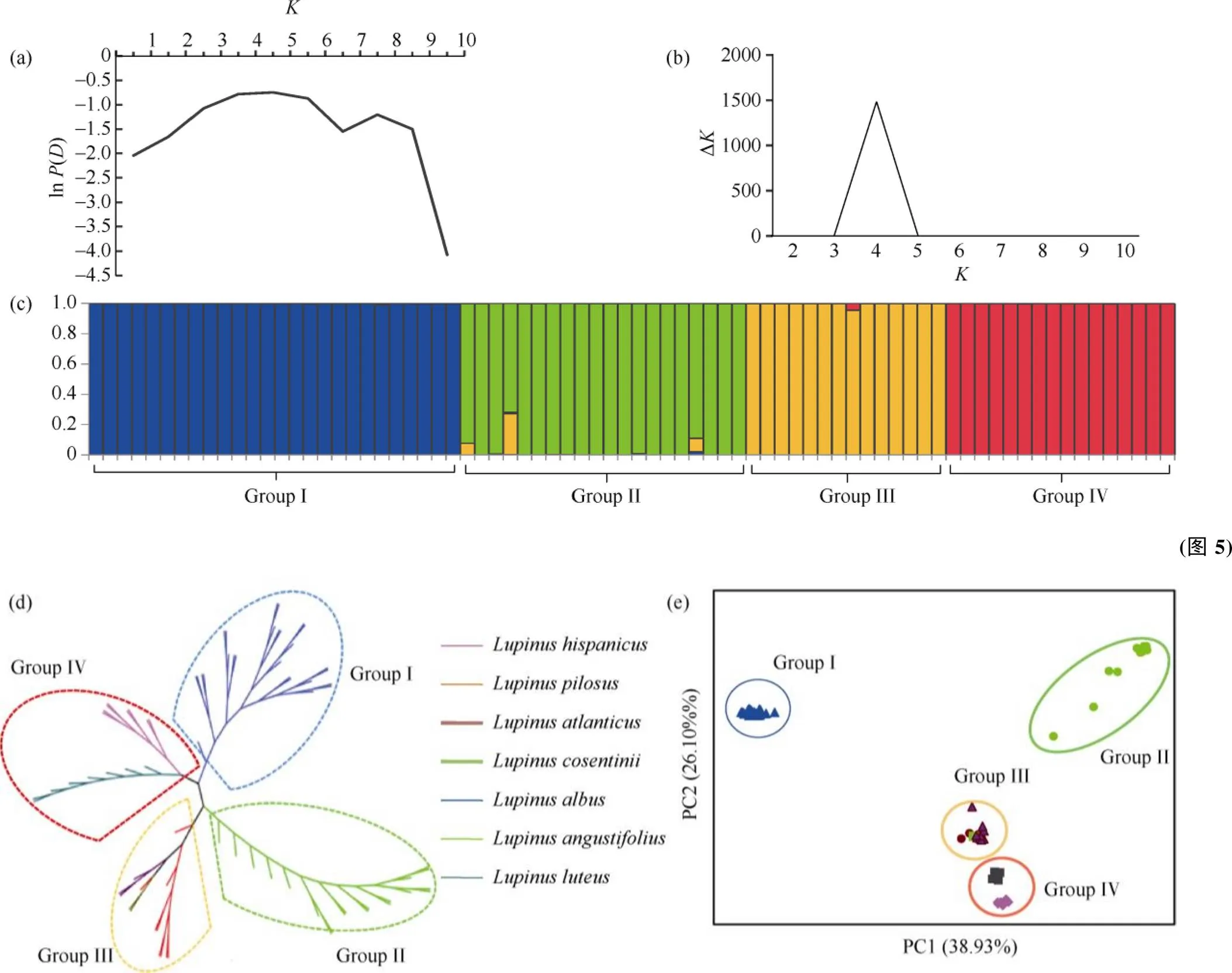
图5 77份羽扇豆资源(“旧世界”)遗传多样性分析
(a)基于ln()平均值, 绘制散点图; (b)基于ln (D)值, 绘制散点图; (c)当= 4时, 77份羽扇豆资源遗传结构; (d)基于Nei’s遗传距离, 77份羽扇豆资源NJ树; (e) 77份羽扇豆资源主成分分析。
(a) the scatter plot of mean ln() from 1 to 10; (b) ln (D) values plotted from 1 to 10; (c) structure of the 77 accessions based on Structure software when= 4; (d) NJ tree of the 77 accessions based on Nei’s genetic distances; (e) principal component analysis of the 77 accessions based on 95 EST-SSR markers.

表3 7个羽扇豆种之间的分子方差分析
Est. Var. 表示具体变异数值; %表示各项变异占总变异的比例;-value表示可信度。
Est. Var. means estimates of variance components; % means percentage of total variance contributed by each component;-value means probability value.
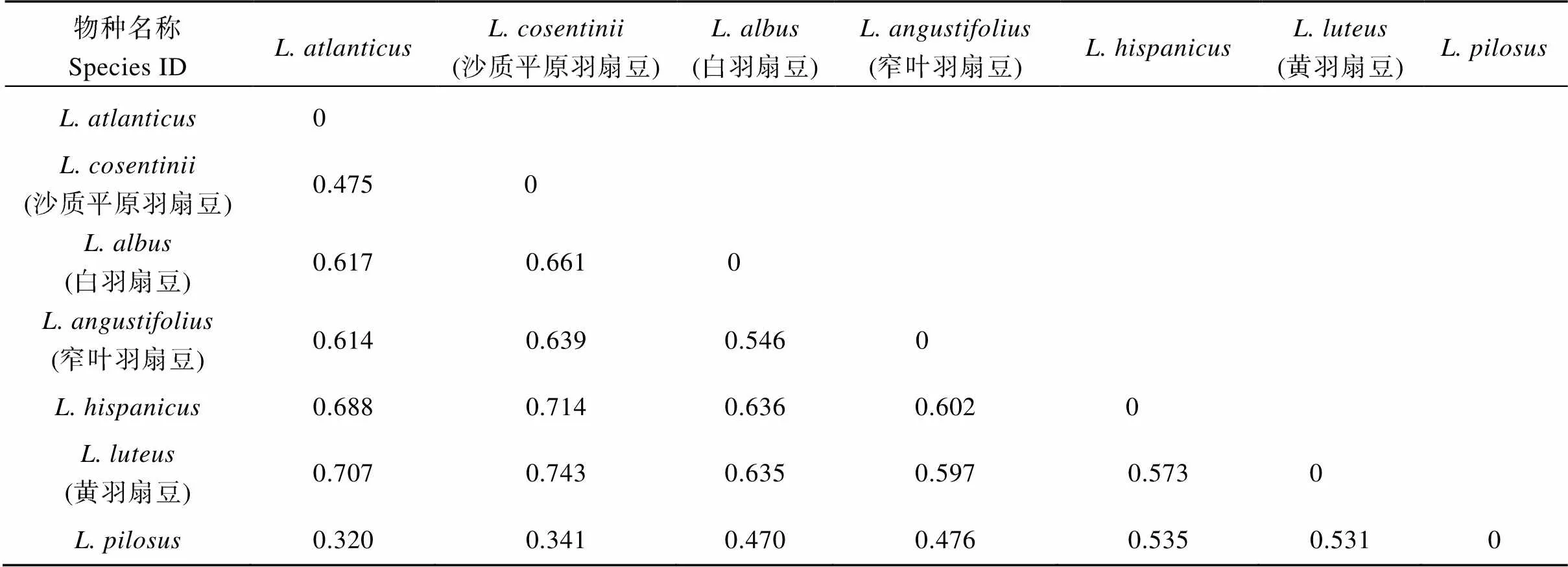
表4 7个羽扇豆种间遗传分化分析
3 讨论
3.1 羽扇豆属分类
羽扇豆属是一个相对较大而多样的属, 一年生或多年生, 主要为草本植物, 少数由柔软的灌木和小乔木组成, 适应广泛的生态地理条件, 世界上约有300个种。核糖体DNA内转录间隔区ITS1和ITS2序列在羽扇豆属内趋于保守, 受到的选择压力较小, 常用于该属较低分类研究。基于rDNA 基因间隔序列(ITS)的研究[23], 羽扇豆属是一个单独的属, 不同种间染色体数目(2= 24到52不等)差异很大。基于种子球蛋白模式、蛋白质血清学、原位杂交技术和基于叶绿体的基因研究[8,24], 羽扇豆属被划分为“新世界” (“New World”)羽扇豆和“旧世界”(Old World)羽扇豆。除依据地理来源(“新世界”-“旧世界”)划分外, 还可以根据叶的特征将羽扇豆分离开来, 一种具有单叶特征(约26个种), 另一种具有典型的掌状叶。通过类黄酮、同工酶、种子球蛋白模式、细胞学、外壳结构比较等研究, 羽扇豆属可被划分为粗糙种子类型和光滑种子类型, 其中“旧世界”主要以光滑种子类型为主, 包括4种类型、、和[25]。本研究通过95对SSR标记对羽扇豆属内22个种系统发育分析表明, 19个羽扇豆种分别被划分到旧世界种群和新世界种群; 另外3个种()未归属到新世界种群, 分析原因可能是本研究选择的羽扇豆种较少或者没有选择外源群, 但整体结果与前人研究结论相对一致。
开展全面、系统的羽扇豆属分类和起源进化的研究, 利用基因组、转录组、叶绿体基因组, 结合形态学数据以及抽取具有更广泛遗传基础的材料, 对我们深入理解羽扇豆属的系统发育重建和进化史具有重要意义。
3.2 旧世界羽扇豆种间遗传多样性分析
种质资源遗传多样性研究是作物遗传改良的重要理论基础。旧世界羽扇豆主要分布在地中海区域、非洲北部和东北部地区, 均为一年生草本植物, 表现为自花授粉, 该地理生态区已成功驯化的栽培种包括白羽扇豆、黄羽扇豆、窄叶羽扇豆和沙质平原羽扇豆。栽培种作为人类和动物重要的植物蛋白质来源和土壤氮肥供给者, 其果实和种子一般都很大, 生物产量较高, 叶子为掌状叶。依据种皮纹理特征可将旧世界羽扇豆分为光滑种子类型和粗糙种子类型2个类群[26]。光滑种子类型包括、、和[25], 主要分布在地中海周围区域, 染色体数目从2= 40到52不等; 粗糙种子类型仅有, 主要分布在北非和地中海地区的东部, 染色体数目从2= 32到42不等。基于ITS序列分析, 可将沙质平原羽扇豆()与和区分开, 但与后者关系更为密切[6]。细胞学研究表明,与遗传关系最近, 与有一定程度的亲缘关系[27-28]。与和的DNA含量非常相似[29], 表明三者遗传关系较为亲近。Talhinas等[30]利用多种分子标记研究了s和-之间的系统发育关系, 表明s与后两者存在明显生殖隔离。Święcicki等[31]培育出和可育种间后代, 说明和亲缘关系较近。
本研究基于95对SSR标记对来源于旧世界的7个羽扇豆种进行遗传多样性分析, 聚类分析、群体结构分析、主成分分析均得到一致的结论, 即7个羽扇豆种的77份材料归属为4个组群(、、和), 与前人研究结果完全一致。
4 结论
22个羽扇豆种分别来源于旧世界和新世界。来源于旧世界的7个羽扇豆种被划分为4个组群(、、和), 各组群所包含的参试资源没有种间交叉重叠。本研究为羽扇豆优异资源的挖掘和创新利用提供理论依据。
附表 请见网络版: 1) 本刊网站http://zwxb.chinacrops. org/; 2) 中国知网http://www.cnki.net/; 3) 万方数据http://c.wanfangdata.com.cn/Periodical-zuowxb.aspx。
[1] 郑卓杰. 中国食用豆类学. 北京: 中国农业出版社, 1997. pp 352–353. Zheng Z J. Food Legumes in China. Beijing: China Agriculture Press, 1997. pp 352–353 (in Chinese).
[2] Eastwood R J, Drummond C S, Schifino-Wittmann M T, Hughes C E. Diversity and evolutionary history of lupins-insights from new phylogenies. In: Palta J A, Berger J B, eds. Proceedings of the 12th international lupin conference. Western Australia: Fremantle, 2008. pp 346–354.
[3] Wolko B, Clements J C, Naganowska B, Nelson M N, Yang H A.. In: Kole C, eds. Wild crop relatives: genomic and breeding resources, legume crops and forages. Berlin: Springer-Verlag, 2011. pp 153–206.
[4] Gladstones J S. Lupins as crop plants., 1970, 23: 123–148.
[5] Wolko B, Weeden N F. Isozyme number as an indicator of phylogeny in., 1990, 31: 179–187.
[6] Aïnouche A, Bayer R J. Phylogenetic relationships in(Fabaceae: Papilionoideae) based on internal transcribed spacer sequences (ITS) of nuclear ribosomal DNA., 1999, 86: 590–607.
[7] Aïnouche A, Bayer R J, Misset M T. Molecular phylogeny, diversification and character evolution in(Fabaceae) with special attention to Mediterranean and African lupines., 2004, 246: 211–222.
[8] Käss E, Wink M. Molecular phylogeny and phylogeography of(Leguminosae) inferred from nucleotide sequences of the rbcL gene and ITS1 + 2 regions of rDNA.,1997, 208: 139–167.
[9] Talhinhas P, Neves-Martins J, Leitao J. AFLP, ISSR and RAPD markers reveal high levels of genetic diversity amongspp., 2010, 122: 507–510.
[10] Sbabou L, Brhada F, Alami I T, Maltouf A F. Genetic diversity of Moroccangermplasm investigated using ISSR and AFLP markers., 2010, 12: 26–32.
[11] Atnaf M, Yao N, Martina K, Dagne K, Wegary D, Tesfaye K. Molecular genetic diversity and population structure of Ethiopian white lupin landraces: Implications for breeding and conservation., 2017, 12: e0188696.
[12] Drummond C S, Eastwood R J, Hughes M C E. Multiple continental radiations and correlates of diversification in(Leguminosae): testing for key innovation with incomplete taxon sampling., 2012, 61: 443–460.
[13] Nevado B, Atchison G W, Hughes C E, Filatov D A. Widespread adaptive evolution during repeated evolutionary radiations in New World lupins., 2016, 7: 12384.
[14] Arenascastro S, Fernándezhaeger J, Jordanobarbudo D. A method for tree-ring analysis using Diva-Gis freeware on scanned core images., 2015, 71: 118–129.
[15] Doyle J J, Doyle J L. A rapid total DNA preparation procedure for fresh plant tissue., 1990, 12: 13–15.
[16] Verhoeven K J F, Jansen J J, Biere D A. Stress-induced DNA methylation changes and their heritability in asexual dandelions., 2010, 185: 1108–1118.
[17] da Maia L C, Palmieri D A, de Souza V Q, Kopp M M, de Carvalho F I F, de Oliveira A C. SSR locator: tool for simple sequence repeat discovery integrated with primer design and PCR simulation., 2008, 2008: 412696.
[18] Liu K, Muse S V. PowerMarker: an integrated analysis environment for genetic marker analysis., 2005, 21: 2128–2129.
[19] Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods., 2011, 28: 2731–2739.
[20] Yeh F C, Boyle T J. Population genetic analysis of co-dominant and dominant markers and quantitative traits., 1997, 129: 157.
[21] Falush D, Stephens M, Pritchard J K. Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies., 2003, 164: 1567–1587.
[22] Peakall R, Smouse P E. GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research: an update., 2012, 28: 2537–2539.
[23] Mahé F, Markova D, Pasquet R, Misset M-T, Aïnouche A. Isolation, phylogeny and evolution of thegene in the legume genusL., 2011, 60: 49–61.
[24] Cristofolini G. A serological contribution to the systematics of the genus(Fabaceae)., 1989, 166: 265–278.
[25] Gladstones J S. Present situation and potential of Mediterranean/African lupins for crop rotation. In: Proceedings of the 3rd international lupin conference. La Rochelle, France, 1984. pp 18–37.
[26] Gladstones J S. Lupins of the Mediterranean region and Africa. South Perth, Australia: Tech Bull, 1974. pp 1–48.
[27] Gupta S, Buirchell B J, Cowling W A. Interspecific reproductive barriers and genomic similarity among the rough-seededspecies., 1996, 115: 123–127.
[28] Gladstones J S. Distribution, origin, taxonomy, history and importance. In: Gladstones J S, Atkins C, Hamblin J, eds. Lupins as Crop Plants: Biology, Production and Utilization. Wallingford, United Kingdom: CAB International, 1998. pp 1–39.
[29] Ghrabi G Z, Puech S, Zouaghi M. Flow cytometry DNA assay of Mediterranean lupins., 1999, 54: 45–56.
[30] Talhinas P, Sreenivasprased S, Neves-Martins J, Oliveira H. Genetic and morphological characterization ofcausing anthracnose of lupins.,2003, 92: 986–996.
[31] Święcickii W K, Święcicki W, Nijaki T.: an interspecific hybrid of Old World lupins., 1999, 68: 217–220.
Assessment of genetic diversity by using EST-SSR markers in
ZHANG Hong-Yan1,5,**, YANG Tao1,**, LIU Rong1,**, JIN Fang2, ZHANG Li-Ke2, YU Hai-Tian3, HU Jin-Guo4, YANG Feng3, WANG Dong1, HE Yu-Hua3,*, and ZONG Xu-Xiao1,*
1Institute of Crop Sciences, Chinese Academy of Agricultural Sciences / The National Key Facility for Crop Gene Resources and Genetic Improvement, Beijing 100081, China;2National Agro-Tech Extension & Service Centre, Beijing 100125, China;3Institute of Food Crops, Yunnan Academy of Agricultural Sciences, Kunming 650205, Yunnan, China;4USDA-ARS Western Regional Plant Introduction Station (WRPIS), Pullman, WA 99164, USA;5Qinghai University, Xining 810016, Qinghai, China
In order to explore the evolutionary relationship ofpreliminarily as well as to excavate and utilize lupin resources from the “Old World” effectively, the genetic diversity among the species undergenus was analyzed. Ninety-five polymorphic pairs of EST-SSR markers developed based on the transcriptome of narrow-leaved lupin (L.) were used to scan 133 lupin accessions from 22 species. A total of 1318 alleles were detected with 13.87 alleles per locus on average, ranging from 3 to 37 alleles; the polymorphism information content (PIC) ranged from 0.39 to 0.91 with the mean value of 0.63; the genetic diversity ranged from 0.41 to 0.92 with the mean value of 0.78. This study showed evolutionary relations among the 22 species undergenus from the “Old World” and the “New World” based on Neighbor-Joining (NJ) method, which is consistent with previous studies. Moreover, seventy-seven lupin accessions of sevenspecies from the “Old World” were divided into 4 groups; there was no overlap of accession from different species contained in each identified group, detected by all the three analysis methods like population structure, cluster analysis based on UPGMA and principal component analysis (PCA).
EST-SSR;genus; transcriptome; genetic diversity
2019-05-21;
2019-09-26;
2019-11-11.
10.3724/SP.J.1006.2020.94077
宗绪晓, E-mail: zongxuxiao@caas.cn; 何玉华, E-mail: trbio@163.com
**同等贡献(Contributed equally to this work)
张红岩, E-mail: 1006248823@qq.com; 杨涛, E-mail: yangtao02@caas.cn; 刘荣, E-mail: liurong@caas.cn
本研究由国家重点研发计划项目(2017YFE0105100), 农作物种质资源保护与利用专项(2019NWB036-07), 中国农业科学院科技创新工程项目和中国农业科学院作物科学研究所中央级公益性科研院所基本科研业务费专项(S2018XC04)资助。
This study was supported by the National Key Research and Development Program of China (2017YFE0105100), the Crop Germplasm Resources Protection (2019NWB036-07), the Agricultural Science and Technology Innovation Program (ASTIP) in CAAS, and the Fundamental Research Funds for Central Non-Profit of Institute of Crop Sciences, CAAS.
URL: http://kns.cnki.net/kcms/detail/11.1809.S.20191108.1915.002.htm
