固相萃取联合超高效液相色谱-串联质谱法测定畜肉中3 种儿茶酚胺类物质
2020-01-07赵文涛吴彦超张颖颖李莹莹郭文萍王守伟
任 南,赵文涛,陈 超*,吴彦超,张颖颖,李莹莹,郭文萍,范 维,王守伟
(中国肉类食品综合研究中心,北京 100068)
儿茶酚胺类是一种含有儿茶酚和胺基的物质,化学结构特点为都有一个双羟基苯核和一个带氨基的侧链[1-2]。肾上腺素、去甲肾上腺素、异丙肾上腺素、肾上腺酮、多巴胺及相关衍生物均属于儿茶酚胺类[3-5]。儿茶酚胺类是体内重要的神经递质,与多种生理、病理现象密切相关,在机体维持内稳态平衡过程中起着关键的作用[6-9]。儿茶酚胺类物质互相之间有着紧密的联系,受多种因素调节[10-12]。自20世纪60年代,肾上腺素类药物一直被认为是增加恢复心跳机会的特效药[13],肾上腺素类药物让人产生巨大的爆发力,为常用急救药物,但过量则可引起中毒,不良反应包括头痛、焦虑不发、烦躁、面色苍白、眩晕、多汗、心跳异常增快等,严重者引发胸痛和心律失常,甚至死亡[14-16];早在1963年,有学者发现,通过使用如咖啡因和肾上腺素等β受体激动类物质,可以促进动物的生长。此后越来越多的β受体激动剂(瘦肉精)被研发出来,后因残留对人体产生危害而被禁止[17-19]。
近年来,注水肉问题受到越来越多的消费者及监管部门的关注。不法分子为了谋取暴利,将儿茶酚胺类药物和水一同注入生猪体内,生猪服用儿茶酚胺类药物后口渴感增强,提高注水量,宰杀后不仅增重而且猪肉颜色鲜红,易于出售[20]。导致近几年注水肉屡禁不止的主要原因是注水肉的关键判定指标不明确,注水肉的生物属性或理化属性研究滞后[21]。我国对猪肉中儿茶酚胺类药物的检测方法鲜见报道,因此研究猪肉中儿茶酚胺类药物的检测方法迫在眉睫,检测方法的建立不仅为探索注水肉的关键指标提供依据,而且为监管部门打击违法行为提供有力手段。
20世纪40年代便有学者对儿茶酚胺进行分析研究,测定方法主要是乙二胺缩合法和三羟基吲哚法[22]。随着新技术的发展,产生了多种测定方法,如化学发光法、荧光光谱法、电化学法、免疫分析法、质谱法以及各种色谱法和多种方法联用等[23-30]。儿茶酚胺的研究多集中于血浆、尿液样本的检测[31-33],肉类样品基质复杂,儿茶酚胺类物质易受到样品的干扰[3],因此如何排除样品的干扰,是畜肉中儿茶酚胺类物质测定的难点。目前儿茶酚胺类物质多用高效液相色谱-电化学法检测,但存在样品前处理时间长、检测时间长、存在干扰等问题[34],而且大多针对体液样品。随着分析技术的不断进步,固相萃取联合液相色谱-串联质谱法应运而生,固相萃取技术是利用选择性吸附和选择性洗脱的液相色谱分离原理,以达到快速分离、提取、富集目标化合物的一种预处理技术。固相萃取技术具有简便、快速、精确、使用溶剂少、回回率高、易于和其他仪器联合使用等优点被广泛应用于多个领域[35]。液相色谱-串联质谱具有检测时间短、样本用量小、灵敏度和特异性高的优点[36]。因此选用固相萃取与液相色谱-串联质谱法,旨在建立一种检测畜肉中儿茶酚胺类药物的灵敏和特异性的方法,并对方法的可靠性进行验证。
1 材料与方法
1.1 材料与试剂
肾上腺素(纯度99.4%) 德国Dr. Ehrenstorfer公司;去甲肾上腺素(纯度98%) 上海阿拉丁公司;异丙肾上腺素标准品(纯度99.0%),乙腈、甲醇(均为色谱纯) 上海发谱实验科技股份有限公司;高氯酸(优级纯)、氨水(分析纯) 国药集团化学试剂有限公司;水无特殊说明均为一级水。
1.2 仪器与设备
1290超高效液相色谱仪、6470三重四极杆串联质谱仪 美国Agilent公司;CR21N离心机 日本Hitachi公司;S-100涡旋仪、SR-IIw振荡器 日本Taiyo公司;ABM-2均质机 株式会社日本精机制作所;Milli-Q纯水仪 美国Millipore公司;Cleanert PCX固相萃取柱博纳艾杰尔公司。
1.3 方法
1.3.1 标准溶液配制
标准储备溶液:准确称取10 mg固体标准品,置于100 mL烧杯中,加适量0.4 mol/L高氯酸溶解,并用0.4 mol/L高氯酸溶液转移并定容至100 mL容量瓶中,摇匀,配制成质量浓度为100 mg/L标准储备液,4 ℃以下冰箱中避光保存。
混合标准溶液:吸取各标准储备溶液适量,用一级水配制成混合标准溶液。
1.3.2 样品提取与净化
提取:称取试样2 g(精确至0.001 g)于100 mL离心管中,加入10 mL 0.4 mol/L高氯酸溶液,15 000 r/min均质2 min,再用10 mL 0.4mol/L高氯酸溶液清洗刀头,12 000 r/min离心15 min,上清液转移至另外一离心管中,残渣再加入10 mL 0.4 mol/L高氯酸溶液,15 000 r/min匀浆2 min进行2 次提取,12 000 r/min离心10 min合并2 次提取溶液,待净化。
净化:Cleanert PCX柱使用前依次用5 mL甲醇、5 mL水、5 mL 0.1 mol/L高氯酸溶液活化,取提取液10 mL至Cleanert PCX柱,以1 mL/min通过PCX小柱后,分别用5 mL 0.1 mol/L高氯酸、5 mL一级水淋洗,最后用5 mL氨水-甲醇溶液(5∶95,V/V)洗脱,回集洗脱液于试管中,40 ℃氮吹近干,用甲酸-水(1∶99,V/V)溶液定容1 mL,涡旋混合1 min,过0.22 μm滤膜后测定。
1.3.3 仪器条件
色谱条件:HILIC Plus色谱柱(4.6 mm×100 mm,3.5 µm);流速0.4 mL/min;流动相A为甲酸-水(1∶99,V/V);流动相B为乙腈;柱温30 ℃;进样量5 μL。洗脱条件见表1。
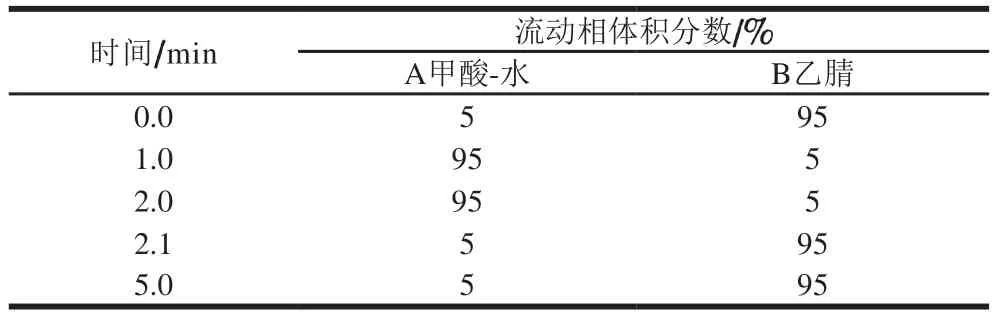
表1 洗脱条件Table 1 Gradient elution program
质谱条件:电喷雾离子源,正离子模式;干燥气温度300 ℃;毛细管电压3 000 V;喷雾器压力35 psi;干燥气流速8 L/min;鞘气流速11 L/min;鞘气温度400 ℃。质谱参数见表2。
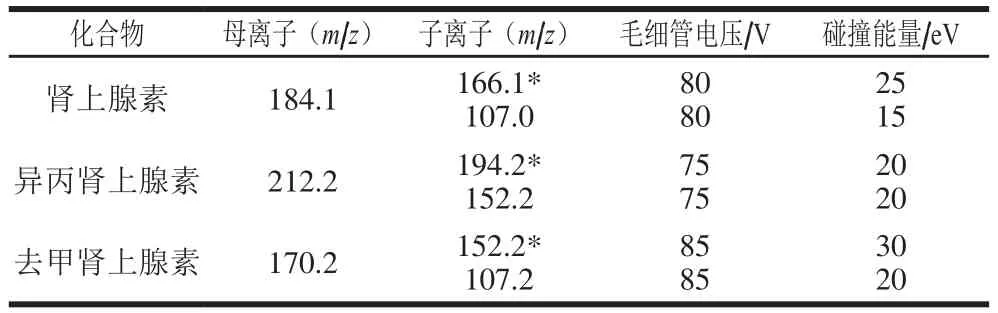
表2 3 种儿茶酚胺类物质的质谱参数Table 2 MS parameters for 3 CAs
2 结果与分析
2.1 质谱条件的优化
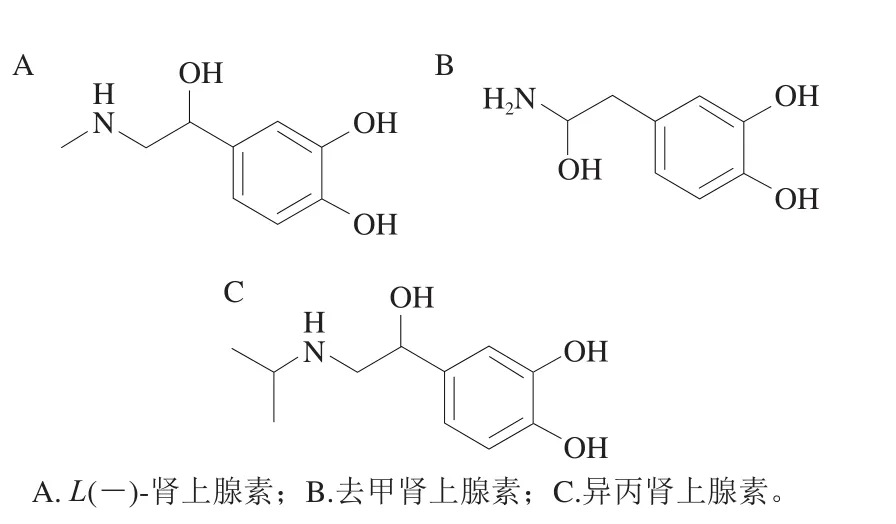
图1 目标化合物结构式Fig. 1 Molecule structures of 3 CAs
如图1所示,3 种化合物均为含氨基的弱碱性化合物,易带正电荷,在正模式扫描下响应值较高,采用选择离子扫描选定化合物母离子,同时优化毛细管出口电压以保证化合物的母离子丰度,选定母离子后再进行子离子扫描,优化碰撞能量确定3 种化合物的子离子(表2)。
3 种化合物结构相似,都带有3 个羟基,均为极性化合物,在非极性色谱柱上保留较弱,为使化合物更好分离和得到良好峰形,本实验选择HILIC亲水色谱柱进行分析,同时在流动相中加入少量甲酸提高样品的离子化效率,以增大化合物响应值,所以本方法选择乙腈与0.1%甲酸-水(1∶99,V/V)作为流动相,结果见图2。
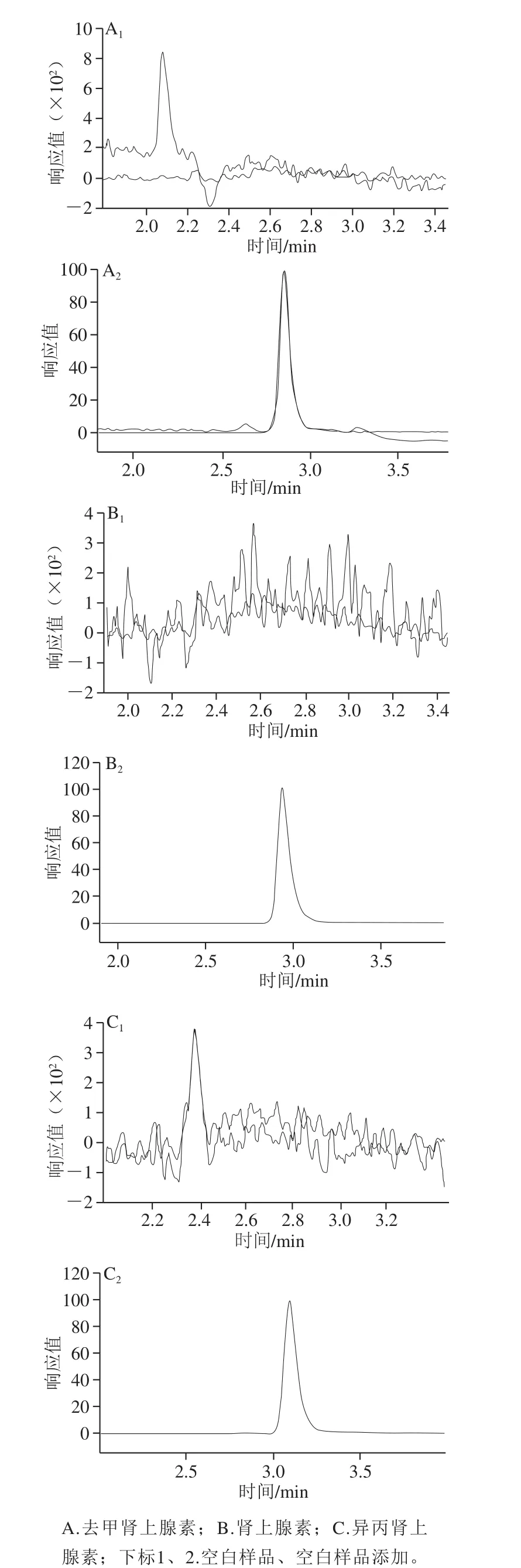
图2 3 种化合物样品空白及样品添加多反应监测图Fig. 2 MRM chromatograms of three CAs in negative and positive samples
2.2 提取溶剂的选择
3 种儿茶酚胺类化合物均含有羟基和氨基,易溶于水溶液和有机溶剂,但由于3 种儿茶酚胺类药物易被氧化,所以在提取溶液中加入适量酸以防止氧化,本实验对比酸化甲醇、酸化乙腈、0.4 mol/L高氯酸溶液、磷酸盐缓冲溶液(pH 4.0)作提取溶剂,加标量为100 μg/kg时的回回率,如图3所示。0.4 mol/L高氯酸溶液作提取溶剂时,3 种儿茶酚胺类均有较高回回率,平均回回率可达80%以上,故选取0.4 mol/L高氯酸溶液作为提取溶剂。
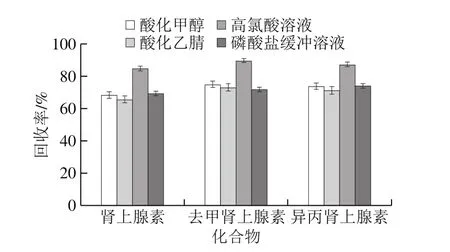
图3 不同提取溶剂回收率对比Fig. 3 Effect of extraction solvents on the recoveries
2.3 提取方式的选择

图4 不同提取方式回收率对比Fig. 4 Effect of extraction methods on the recoveries (n = 3)
肉类由于其脂肪、蛋白含量比较高,某些基质会对样品提取的过程造成影响[37],因此考察常见4 种提取方式,结果见图4。匀浆不仅误差较小而且提取效率可达到85%以上,因此选用匀浆为提取方法。
2.4 固相萃取柱的选择
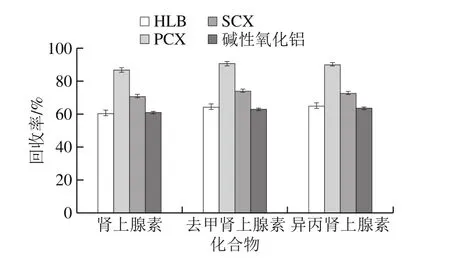
图5 不同固相萃取柱回收率对比Fig. 5 Effect of SPE columns on the recoveries
考察HLB柱、PCX柱、SCX柱、碱性氧化铝柱对目标化合物的提取、净化效果,结果见图5。固相萃取柱对于3 种目标物质的保留都比较稳定,PCX柱对3 种目标化合物(添加量40 μg/kg)回回率均高于其他固相萃取柱。
从提取溶剂、提取方式、固相萃取柱的选择看,3 种化合物之间回回率具有相似的规律,这可能是由于它们的结构和化学性质相似。肾上腺素在不同的影响因素中回回率最低,可能是其化学性质与另外2 种相比更不稳定,更容易被外界(基质/环境)因素干扰。不同提取溶剂和不同固相萃取柱对3 种化合物内回回率的误差差异较小,不同提取方式对3 种化合物内回回率的误差差异较大,因此提取方式的选择对方法的稳定性影响较大。
2.5 基质效应评价
基质效应是由基质中的共提干扰物(非目标化合物)与目标化合物竞争电离所致[38]。基质效应会影响检测结果的准确性,因此需要进行基质效应的评价[39]。实验选取猪肉、牛肉、羊肉进行基质效应评价,结果见表3,3 种药物在猪肉、牛肉、羊肉基质中都属于强基质效应,如果用溶剂配制标准曲线定量,会造成结果有偏差,因此需要选择基质标准曲线进行定量,以消除基质效应的影响。

表3 3 种药物的基质效应评价Table 3 Matrix effect evaluation of three CAs
2.6 方法学结果
2.6.1 标准曲线、检出限与定量限测定结果
用空白基质配制标准溶液,制作标准曲线,质量浓度分别为40、50、100、500、1 000 ng/mL,在选定的前处理和仪器条件下进行测定,得到不同质量浓度的色谱图,以添加儿茶酚胺类药物质量浓度为横坐标,儿茶酚胺类药物峰面积为纵坐标,得到3 种化合物的线性方程及相关系数。利用逐级稀释法方空白样品中添加已知质量浓度的混合标准溶液,以3 倍信噪比确定检出限,10 倍信噪比确定定量限,结果见表4。该方法在40~1 000 μg/kg范围内3 种儿茶酚胺类物质的线性良好,相关系数均大于0.99,检出限为10 μg/kg,定量限为40 μg/kg。
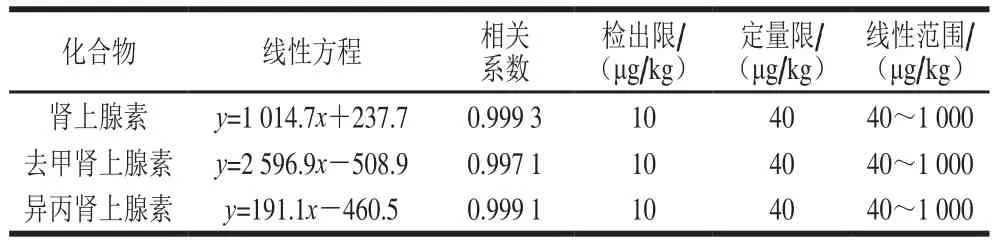
表4 3 种儿茶酚胺类药物的线性方程、相关系数、检出限、定量限、线性范围Table 4 Regression equations, correlation coefficients, LODs, LOQs and linear ranges of three CAs
2.6.2 加标回回率及相对标准偏差测定结果
分别称取牛肉、猪肉、羊肉空白样品各18 份,每份2.0 g,每种空白样中分别添加40、400、1 000 μg/kg 3 个水平的混合标准溶液,每个水平6 个平行样品,后按1.3.2节和1.3.3节方法制备进行实验,结果见表5。不同添加水平的3 种儿茶酚胺类物质的回回率在75.48%~91.25%之间,相对标准偏差为1.2%~4.6%,回回率和相对标准偏差均符合日常检测的要求。

表5 猪肉、牛肉、羊肉中加标回收率及相对标准偏差结果Table 5 Spiked recoveries for three CAs in different matrix samples(n= 6)
2.7 标准溶液稳定性结果
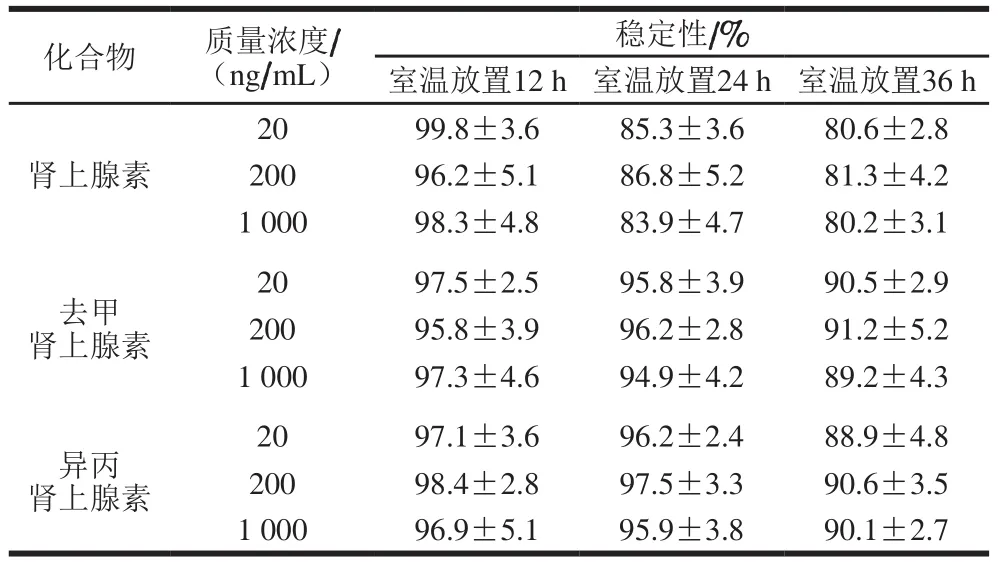
表6 放置不同时间3 种药物标准溶液的稳定性Table 6 Storage stability of standard solutions of three CAs
将低、中、高,3 个质量浓度的肾上腺素、去甲肾上腺素、异丙肾上腺素标准溶液混标放置于室温(25 ℃)下,比较同一条件下不同放置时间的标准溶液稳定性差异。如表6所示,3 种药物在室温放置12 h较稳定,放置24 h后肾上腺素开始逐渐降解,质量浓度降低至原来的85%,放置36 h后肾上腺素质量浓度降低至原来的80%,去甲肾上腺素和异丙肾上腺素质量浓度降低至原来的90%,所以为了保证结果准确,建议标准溶液临用现配,同时样品提取液需在12 h内完成上机检测。
2.8 实际样品的测定结果
采用本方法对市售的140 件畜肉进行测定,种类包括牛肉、羊肉、猪肉,1 件猪肉样品检出肾上腺素,含量为50 μg/kg。同时对该样品进行水分测定,结果大于77%,不符合GB 18394—2001《畜禽肉水分限量》的要求,表明儿茶酚胺类物质对水分是否超标具有一定的指示作用。
3 讨 论
本实验采用固相萃取技术与超高效液相色谱-串联质谱法同时测定畜肉中肾上腺素、去甲肾上腺素、异丙肾上腺素3 种儿茶酚胺类药物,方法简便、快速。方法学评价结果表明,本方法准确、可靠,为考察注水肉的关键指标提供思路,为监管部门打击违法行为提供手段。同时方法还存在一些不足,对于同种影响因素内部误差产生的差异以及不同化合物间回回率差异的原因仍需进行大量研究工作。儿茶酚胺类化合物大部分是生物体内正常代谢产生的物质,因此如何判定外源性注射儿茶酚胺仍需进一步研究。
