双靶向配体化力达霉素在大鼠体内药动学及组织分布和排泄
2019-12-31宋文凭张玉民樊慧蓉刘鉴峰邵荣光
叶 程,曹 睿,宋文凭,张玉民,樊慧蓉,刘鉴峰,李 亮,邵荣光
(1.中国医学科学院医药生物技术研究所,北京100050;2.中国医学科学院放射医学研究所,天津300192)
双靶向配体化力达霉素(dual targeting ligandbased lidamycin,DTLL)是在强效抗肿瘤抗生素力达霉素(又称C-1027)[1-2]辅基蛋白结构基础上,通过生物工程技术改造得到的,具有靶向表皮生长因子受体和人类表皮生长因子受体2功能的力达霉素衍生物,其相对分子质量约19 000[3]。实验证明,DTLL在多种肿瘤细胞和动物模型中均表现出良好的抗肿瘤活性,较之力达霉素可以显著提升对肿瘤细胞的杀伤,同时,制备工艺成熟稳定,具有良好的应用前景[4-6]。因此,观察DTLL在动物体内的药动学特征及分布和排泄数据,对该药的后续研究和临床试验申报具有重大参考意义。
放射性同位素示踪法是将蛋白质类药物通过同位素标记后给予实验动物,用以检测药物在实验动物体内的血药浓度变化以及分布代谢情况的方法,具有灵敏度高、操作简便等特点[7]。本研究中采用的放射性同位素125I具有半衰期适中、易于标记和检测等优点,因此被广泛应用于蛋白质类药物的体内药动学研究。本研究将[125I]DTLL iv 给予大鼠后,检测血浆中放射性浓度并计算药动学参数,进一步分析DTLL 在大鼠体内的分布和排泄,为下一步研究奠定实验基础。
1 材料与方法
1.1 动物、试剂和仪器
健康Wistar 大鼠,雌性170~260 g,雄性280~300 g,SPF级(北京维通利华实验动物技术有限公司)。中国医学科学院医药生物技术研究所制备的DTLL 冻干粉针,批号P10327 和P20327,纯度>95%;其余试剂均为国产分析纯。2470型γ计数仪(美国PerkinElmer 公司);Ultimate3000 型高效液相色谱(美国Dionex 公司);Mini-scan TLC 薄层放射性扫描仪(美国BIOSCAN公司)。
1.2 [125I]DTLL的制备
采用氯胺T 法将125I 标记于DTLL 蛋白上,经Sephadex G-50 凝胶纯化,洗脱液为50 mmol·L-1PBS(pH7.4),连续收集[125I]DTLL峰洗脱液。
1.3 标记率的测定
在硅胶板的初始端点2 μL标记样品进行层析,展开剂为乙醇∶水=9∶1(V/V)。至距离硅胶板末端约2 cm处停止,利用薄层放射性扫描仪检测标记物的标记率。
1.4 放射化学纯度测定
分别收集纯化样品2 μL 进行层析,展开剂为乙醇∶水=9∶1(V/V)。至距离硅胶板末端约2 cm处停止,利用薄层放射性扫描仪检测纯化后标记物的放射化学纯度。
1.5 血清样品制备
健康Wistar 大鼠6 只,雌雄各半,尾静脉注射[125I]DTLL,给药剂量2625 kBq·0.05 mg-1·kg-1。于iv 给药后0.083,0.25,0.5,1,1.5,2,3,4,6,8,12 和24 h,从眼眶后静脉丛取血约300 μL,离心分离血清。吸取血清样品100 μL置于离心管中,加入三氯醋酸400 μL,振荡摇匀,离心后分别取上清液以及下层沉淀,测定放射性活度。
1.6 组织分布测定
健康Wistar 大鼠24 只,雌雄各半,随机分成4 组,每只大鼠尾静脉注射[125I]DTLL,给药剂量2625 kBq·0.05 mg-1·kg-1。分别于给药后的4个时间点(0.083,0.5,3 和6 h)眼眶后静脉丛取血并处死大鼠,立即解剖,分别取心、肝、脾、肺、肾、胃、肠、脑、肌肉、脂肪、生殖腺和淋巴结。将上述组织称重,用生理盐水充分浸泡后剪碎制成匀浆,取100 μL匀浆加入三氯醋酸400 μL,振荡摇匀,离心后分别取上清液以及下层沉淀,测定放射性活度。
1.7 粪便和尿液排泄测定
健康Wistar大鼠12只,雌雄各半,分成2组,给药前自由进食与饮水,每只大鼠尾静脉注射[125I]DTLL,给药剂量2625 kBq·0.05 mg-1·kg-1。将大鼠置于代谢笼中,分别收集给药后0~3,3~5,5~7,7~11,11~24,24~48,48~72,72~96,96~120,120~144 和144~168 h 各时间段内的尿样及粪样,记录尿样体积,称粪便质量,直接测定放射性活度。
1.8 胆汁排泄测定
取健康Wistar大鼠6只,雌雄各半,给药前自由进食与饮水。大鼠腹腔注射20%乌拉坦麻醉,实施胆汁插管引流手术后,每只大鼠尾静脉注射[125I]DTLL,给药剂量2625 kBq·0.05 mg-1·kg-1。分别于给药后0~1,1~2,2~3,3~4,4~5,5~6,6~7,7~8,8~9,9~10,10~11,11~12和12~24 h收集全部胆汁样品,并直接测定放射性活度。计算胆汁中药物的排泄量、累积排泄量和累积排泄分数。
2 结果
2.1 [125I]DTLL放射化学纯度和标记率
将125I 标记DTLL 后的混合液,经Sephadex G-50 凝胶纯化得到[125I]DTLL(图1),标记率为92.52%(图2)。根据薄层放射性扫描仪的测定结果和积分结果,测得第9~12 管纯化后的[125I]DTLL 的放射化学纯度均>99%,且混合后[125I]DTLL 的放射化学纯度为98%,满足后续实验需求(图3)。样品总放射性浓度为2.35×107kBq·L-1,蛋白质浓度为447.7 mg·L-1,比放射性活度(总放射性浓度/蛋白质浓度)为5.25×107kBq·g-1蛋白。
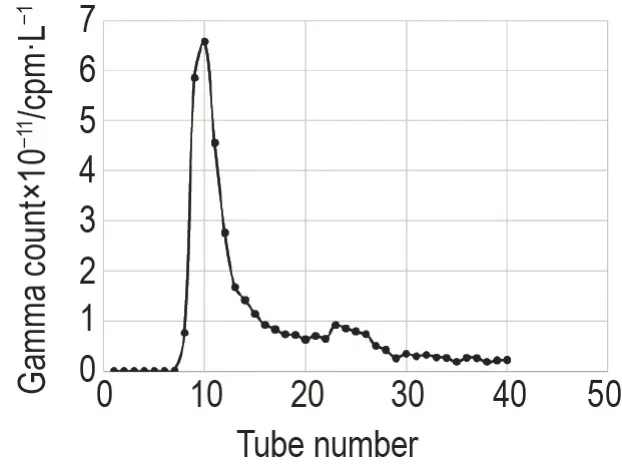
Fig.1 Purification of125I-labeled dual target ligandbased lidamycin([125I]DTLL)by Sephadex G-50 column.Reaction mixture of [125I] DTLL was transferred to the Sephadex G-50 column for purification. The eluent was PBS (50 mmol·L-1,pH 7.4). The flow rate was 1.0 mL·min- 1. The radioactivity of effluent was detected in every each milliliter.

Fig.2 Detection of isotope labeling yield of[125I]DTLL by thin-layer chromatograph. Labeling yield was calculated according to the determination results and integral results of thin-layer radioactive scanner.
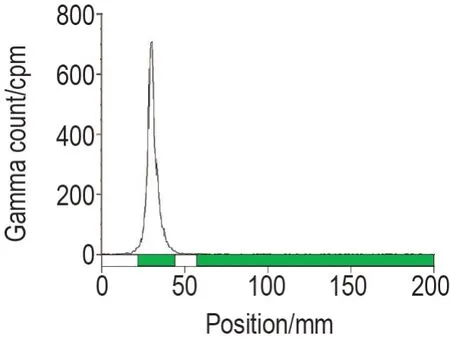
Fig.3 Detection of radiochemical purity of[125I]DTLL by thin-layer chromatograph. Radiochemical purity was calculated according to the determination results and integral results of thin-layer radioactive scanner.
2.2 大鼠尾静脉注射[125I]DTLL后的药动学参数
大鼠尾静脉给药后,通过测定的三氯醋酸沉淀放射性和总放射性,分别计算对应的药物浓度。结果表明,三氯醋酸沉淀放射性和总放射性药物浓度变化趋势一致,均随着给药时间的增加而逐渐降低,但总放射性计算得到的药物浓度远高于三氯醋酸沉淀放射性计算得到的药物浓度(图4)。分别以总放射性与三氯醋酸沉淀放射性计算[125I]DTLL的浓度-时间数据,并通过药动学软件以非房室模型计算参数(表1)。在主要药动学参数中,与体内暴露量密切相关的药动学参数如AUC0-t,AUC0-∞,MRT0-t和t1/2的总放射性均高于酸沉放射性,而与体内分布和清除相关的Vd和Cl 则是三氯醋酸沉淀放射性高于总放射性。

Fig.4 Concentration-time curves of[125I]DTLL in plasma of rats after iv administration. Wistar rats were iv administred with[125I]DTLL 0.05 mg·kg-1.The blood sample was collected before administration and after 0.083,0.25,0.5,1,1.5,2,3,4,6,8,12 and 24 h of administration. Total radio-activity was determined by LSC.[125I]DTLL concentration(ng·Eq·mL-1)=Measured radioactivity/decay coefficient/total radioactivity of[125I]DTLL solution/0.1×1000.,n=6.
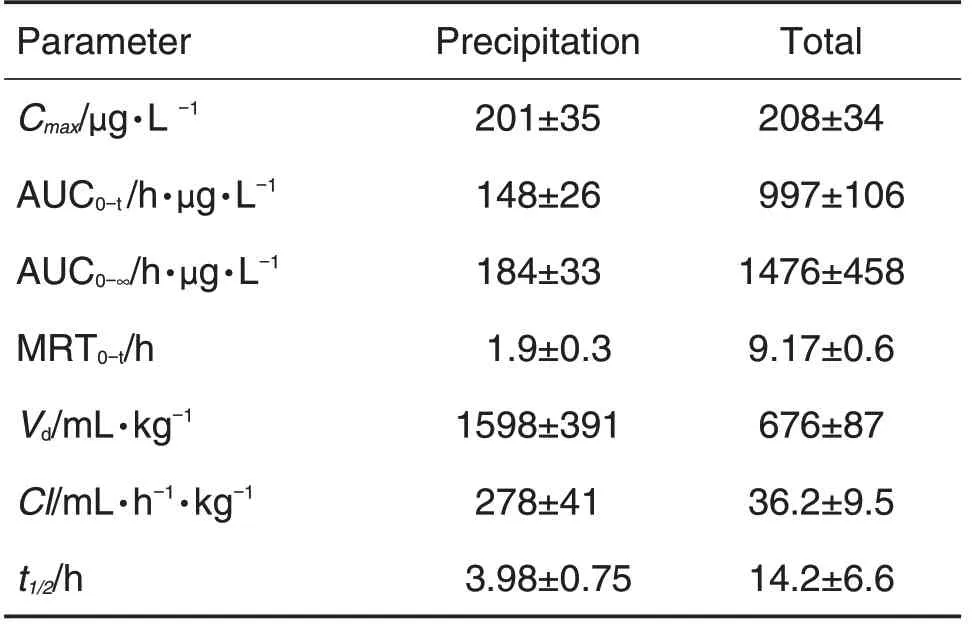
Tab.1 Pharmacokinetic parameters of [125I] DTLL in rats after iv administration
2.3 大鼠尾静脉注射[125I]DTLL 后的全身组织放射性分布
大鼠尾静脉注射[125I]DTLL 后对各组织/血清中放射性进行分析比较。结果表明,大鼠尾静脉注射给药后,酸沉部分和总放射部分放射性在大鼠体内主要脏器的分布范围基本一致,广泛分布于各组织中,但在血液以及肺、肝等血流丰富的脏器中,总放射性浓度均高于酸沉部分放射性浓度,表明对应组织的放射性可能是由血液中游离125I 造成的(图5);在比较0~6 h 内主要脏器的酸沉部分药物浓度后发现,DTLL 在主要脏器中均未随时间延长发生蓄积的情况,其中大部分脏器给药后放射性分布逐渐降低,而心脏和胃壁在给药后3 h 达到峰值(表2)。
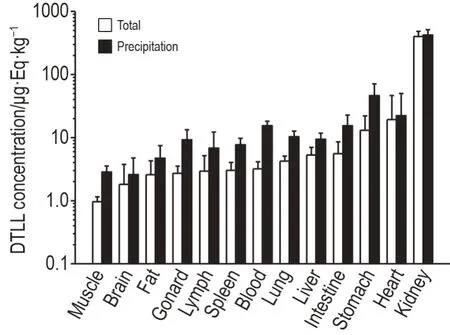
Fig.5 Tissue distribution of[125I]DTLL in rats 6 h after iv administration. See Fig.4 for the rat treatment. Tissues were collected after 6 h of administration.[125I]DTLL concentration(μg·Eq·kg-1)=Measured radioactivity/decay coefficient/0.7/30/tissue mass/(total radioactivity of DTLL solution/0.7/30/1000).,n=6.
2.4 大鼠尾静脉注射[125I]DTLL后的放射性排泄
大鼠iv 给予[125I]DTLL 后绘制累积排泄曲线。在168 h 内经尿液排泄的累积排泄分数为(82.5±2.6)%,经粪便排出的累积排泄分数为(10.4±6.6)%,累积尿粪排泄的放射量达到给药剂量的(92.9±7.4)%(图6),而0~24 h 内经胆汁排泄的放射剂量仅为总剂量的(3.37±2.1)%(图7)。以上结果表明,粪便和尿液是[125I]DTLL 主要的排泄途径。
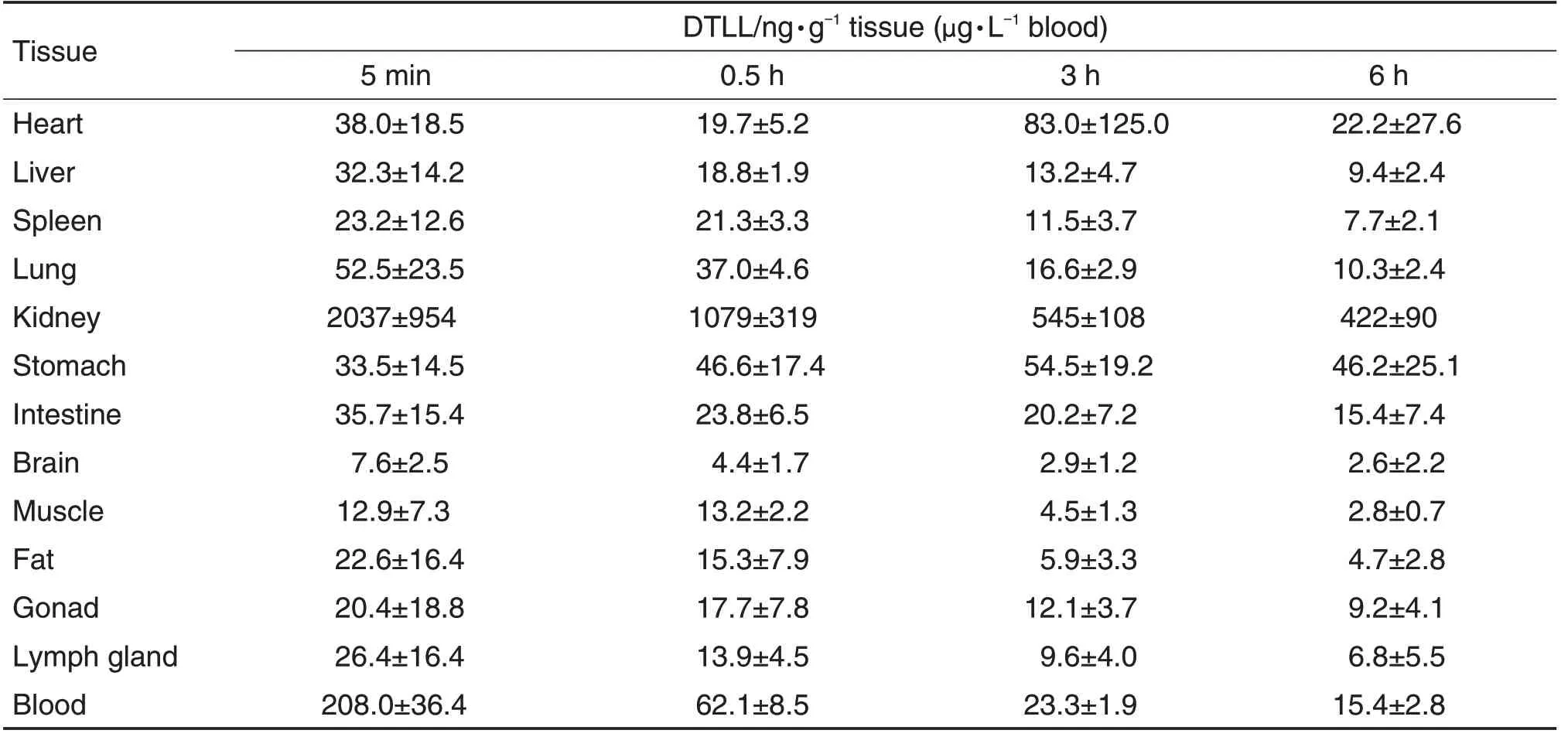
Tab.2 Tissue distribution of DTLL in rats after iv administration
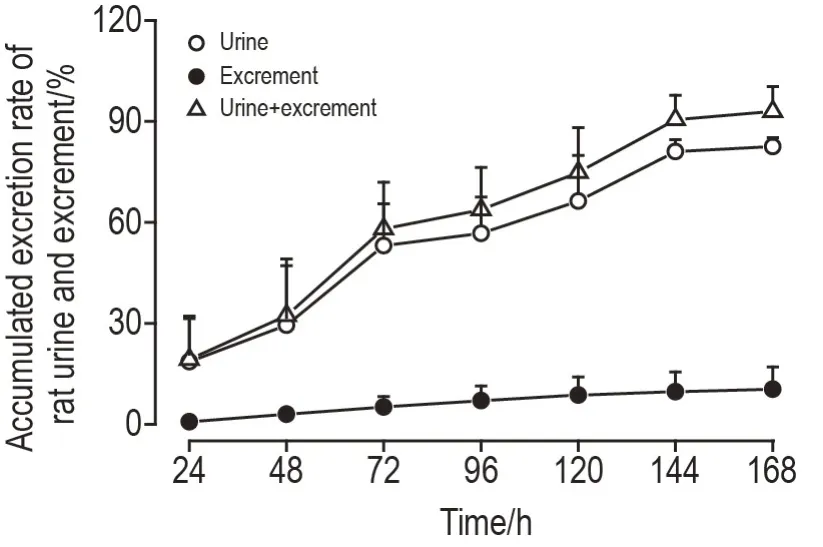
Fig.6 Accumulated excretion curves of rats urine and excrement after[125I]DTLL administration. See Fig.4 for the rat treatment. Urine and feces were collected at 0-3,3-5,5-7,7-11,11-24,24-48,48-72,72-96,96-120,120-144 and 144-168 h after administration.Urine excretion amount=radioactivity of 1 mL urine×urine volume(cpm);feces excretion amount=total measured solid radioactivity(cpm). Biles were collected at 0-1,1-2,2-3,3-4,4-5,5-6,6-7,7-8,8-9,9-10,10-11,11-12 and 12-24 h after administration.,n=6.
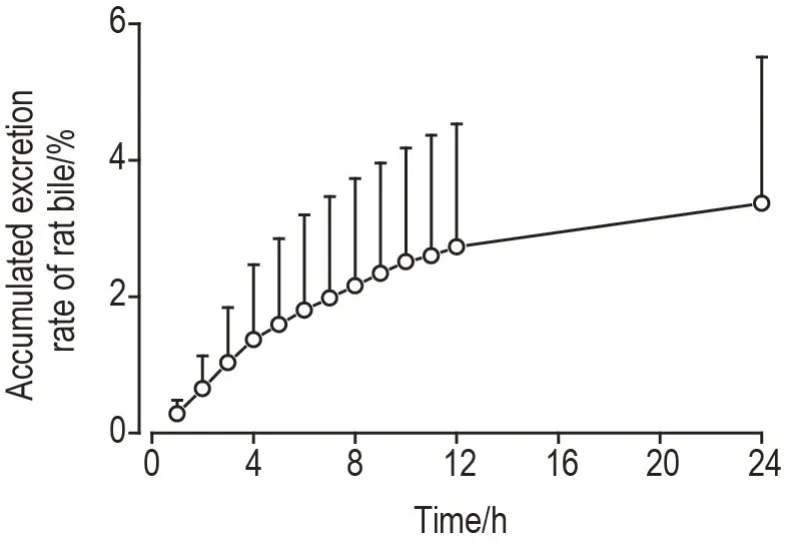
Fig.7 Accumulated excretion curve of rat bile after[125I]DTLL administration. See Fig.4 for the rat treatment.Biles were collected at 0-1,1-2,2-3,3-4,4-5,5-6,6-7,7-8,8-9,9-10,10-11,11-12 and 12-24 h after administration.Excretion rate(%)=excretion amount/dosage×100%.,n=6.
3 讨论
大分子药物进入体内后的分布和排泄过程通常比较复杂,通常会以降解产物的形式,经肝肾代谢等途径排出体外。因此,针对大分子药动学研究所选的方法,应能将进入体液的药物及其降解、代谢产物与内源性物质区别开来,以排除干扰物等可能产生的干扰[8]。放射性同位素125I 具有半衰期适中,易于标记和检测等优点,既可以避免血液中杂质对结果测定的干扰,同时又可以简化样品处理和检测过程、缩短实验周期。因此,在蛋白类药物的药动学研究中具有独特的优势。
125I 标记的外源性蛋白或多肽在进入体内生理条件的影响下后可能被降解脱碘,因此直接检测血液中放射性活度并不能准确反映药物的血药浓度。本研究首先比较了血清总放射性和三氯醋酸沉淀放射性的差异,通过比较,将血清样品中的[125I]DTLL 与分解游离的125I 区分开。结果显示,[125I]DTLL 进入体内后很可能被降解并释放出游离125I。因此,以三氯醋酸沉淀组放射性值计算得到的数值理论上更接近实际情况。值得注意的是,大分子生物制品的表观分布容积Vd与血浆容积相近,如贝伐珠单抗、英利昔单抗和利多昔单抗等,单剂量的(Vd)均在10~30 mL·kg-1之间;而本研究中大鼠iv给予[125I]DTLL 后的药物Vd值达到了(1598±391)mL·kg-1,推测其原因可能是DTLL不会与血浆蛋白或其他非特异性靶标结合,但能与靶标组织特异性的结合,导致其Vd相对较大。
刘有平等[9]曾对小鼠静脉注射力达霉素的体内血清动力学进行考察。在同等给药剂量下,经三氯醋酸沉淀处理后,力达霉素原型药物的AUC0-24h为57.5 h·μg·L-1,t1/2为2.78 h。将力达霉素药动学结果与本研究的大鼠iv 给予[125I]DTLL 的结果相比,相对应的参数差异不大。而且Richter 等[10]证实,生物大分子药物的药动学特征相对单一,个体差异小,易于进行种属外推和药动学参数预测。因此推测,力达霉素经过结构改造后得到的DTLL 的药动学特征未发生明显改变。当然,由于生物大分子药物存在抗原性,为进一步观察其不同种属间差异,后续仍需采用非啮齿类动物(如犬、猪或恒河猴等)对DTLL的药动学特征加以深入研究。
而[125I]DTLL 在动物体内分布研究证明,经iv给药后,[125I]DTLL 在肺、肾和心脏等组织中分布含量较高,与上述表征分布特征的药动学参数的结果相吻合。而在给药72 h后,肾和心脏中分布仍能保持在较高水平。其中肾的药物浓度与其余组织相比高出数倍,但随着时间的推进,肾中药物浓度呈下降趋势,表明DTLL可以通过肾消除,但不会在肾中蓄积;心脏中的药物浓度随时间不同呈不规律分布,但因浓度较低,因此也可排除其蓄积趋势;其余组织均呈下降趋势且含量较低,可以推定不会在这些组织中引起蓄积;脑中药物含量极低,提示可能血脑屏障阻碍了DTLL进入脑部。
大分子药物通常不会以原型药物形式排泄出体外,而是在体内环境中被进一步降解代谢为氨基酸、多肽或小分子产物,经肾滤过或胆汁排泄出体外[11]。因此本研究采用测定总放射性来估算DTLL及其降解产物的总排泄量。排泄研究结果显示,168 h内经尿液和粪便的排泄量达给总给药剂量的(92.9±7.4)%,而从胆汁排出的放射性仅占给药剂量的(3.4±2.1)%。表明DTLL 主要从肾排泄,其次为肠道排泄。上述结果提示,经iv给药后,DTLL在大鼠体内可被快速消除,无蓄积风险,为DTLL后续研究提供数据支持。
