生物分离法制备细胞色素C质量的影响因素
2019-12-30刘文静郑永祥唐章勇
刘文静,郑永祥,张 革,唐章勇,余 蓉
(1.四川大学 华西药学院,四川 成都 610041;2.四川德博尔制药有限公司,四川 德阳 618000)
细胞色素C是由103~113个氨基酸残基组成、分子量约为12 000~13 000的一种重要的氧化还原血红蛋白酶,是生命体内氧化还原链上重要的电子传递体。细胞色素C肽链中心包裹着一个铁卟啉环,通过铁卟啉环中心Fe价态的转换来达到纠正呼吸的作用[1]。目前,细胞色素C在临床上主要用于各种组织缺氧的急救;对抗癌药物引起的白细胞降低、四肢循环障碍、肝脏疾患和肾炎亦有一定改善作用[2];同时有研究发现,细胞色素C可诱导细胞凋亡,用于抗癌治疗[3-5]。
但细胞色素C存在稳定性不好、诱发临床过敏等问题[6-7],限制了其在临床上的广泛应用。而过敏反应的发生与纯度有关,纯度越高,过敏发生率越低[8]。为了减少细胞色素C临床副反应,提高药物疗效,进一步提高细胞色素C质量,需严格控制细胞色素C生产工艺。生化药品的质量受到生产过程各个环节因素的影响,细胞色素C传统生产工艺常用三氯乙酸作为沉淀剂,通过可逆沉淀来纯化细胞色素C,然而有研究报道三氯乙酸可能会引起细胞色素C发生变性与聚合[9]。因此,目前有研究者对细胞色素C传统生产工艺进行了改进[10-11],并对新旧工艺制备的细胞色素C纯度进行分析[12]。但对于传统工艺中三氯乙酸如何影响细胞色素C的质量尚未明确阐明,深入研究三氯乙酸对细胞色素C质量的影响及其作用方式有助于为细胞色素C生产工艺改进提供理论指导。
本文中,笔者采用十二烷基硫酸钠-聚丙烯酰胺凝胶电泳法(SDS-PAGE)、液相色谱-串联质谱法(LC-MS/MS)来对比分析三氯乙酸对细胞色素C质量的影响,以期提高细胞色素C质量。
1 材料与方法
1.1 材料
1.1.1 供试样品
传统工艺制备的细胞色素C原料药溶液(批号:2018001)、新工艺制备的细胞色素C原料药溶液(批号:2018002、2018003、2018004、2018005、2018006、2018007),四川德博尔制药有限公司;新工艺制备的细胞色素C原料药溶液(批号:2018008),实验室自制;细胞色素C对照品(细胞色素C注射液,批号:20170422),安徽宏业药业有限公司;细胞色素C标准品(批号:140670-200501),中国食品药品检定研究院。
1.1.2 试剂与仪器
乙腈(色谱级)、甲酸(色谱级)、甲苯磺酰苯丙氨酰氯甲酮(TPCK)处理的胰蛋白酶(T1426)、三羟甲基氨基甲烷,Sigma-Aldrich公司;预染蛋白(Marker),美国Thermo Scientific公司;其余试剂皆为市售国产分析纯。
液相色谱-四级杆飞行时间串联质谱(miroTOF-QⅡ质谱仪,Bruker Daltonics公司;LC-20AD型纳升液相仪,Shimadzu公司);Sorvall ST 16R型冷冻离心机、902-ULTS型酶标仪,Thermo Scientific公司;VE-186型电泳系统,上海Tanon公司;2014AE64型全自动凝胶成像系统,北京五洲东方科技发展有限公司;UPH-I-20T型超纯水器,四川优普公司;丁基-琼脂糖凝胶(Butyl-Sepharose FF)层析柱,常州天地人和公司;HD-21-1 型核酸蛋白检测仪,HD-A型电脑采集器,上海青浦沪西仪器厂。
1.2 方法
1.2.1 细胞色素C制备
细胞色素C的传统工艺参照文献[13]进行。
细胞色素C新工艺[12]采用疏水层析纯化取代了三氯乙酸沉淀,其他步骤都应用传统工艺[13]。
疏水层析纯化:盐析后细胞色素C 上清液用丁基-琼脂糖凝胶(Butyl-Sepharose FF)疏水层析柱纯化,采用硫酸铵浓度梯度洗脱。A洗脱液为含质量分数为45%硫酸铵的20 mmol/L磷酸盐(pH 7.0)缓冲液;B洗脱液为20 mmol/L磷酸盐(pH 7.0)缓冲液。洗脱程序为0~40 min 100%A,40~80 min 25%B,80~120 min 50%B,120~160 min 75%B,160~200 min 100%B。先用100%A洗脱液平衡柱子,将盐析后含细胞色素C 的上清液缓慢加入层析柱中,按照上述程序进行梯度洗脱,每个梯度用约10倍柱体积(柱体积5 mL)洗脱液洗脱。流速1.2 mL/min,检测波长280 nm,收集最大吸收峰洗脱液。洗脱液经超滤,透析得到新工艺制备的细胞色素C原料药溶液样品(批号:2018008)。
制备的细胞色素C样品经过2015版《中国药典》[14]细胞色素C溶液项下的鉴别、含量测定,符合药典规定。
1.2.2 SDS-PAGE分析细胞色素C
非还原SDS-PAGE法分析细胞色素C纯度:配制5%浓缩胶与15%分离胶的不连续缓冲系统。细胞色素C原料药样品、对照品、标准品分别加超纯水稀释至1 mg/mL,按体积比4∶ 1分别加入5倍非还原蛋白上样缓冲液,混匀,在沸水中孵育5 min,离心。细胞色素C样品上样量6 μL、标准蛋白上样量3 μL。电泳操作参见2015版《中国药典》通则0541第五法[15]。
还原性SDS-PAGE:取含有分子量2.5×104蛋白的细胞色素C标准品、对照品、原料药样品(2018001批次)进行还原性SDS-PAGE,以上3批样品分别加超纯水稀释至1 mg/mL,按体积比4∶ 1分别加入5倍还原性蛋白上样缓冲液处理,其余实验操作同上述非还原SDS-PAGE法。
1.2.3 LC-MS/MS分析与鉴定细胞色素C二聚体
色谱条件:Acclaim PepMap 100 C18 (5 μm,5 mm×0.3 mm,10 nm)预柱,Thermo Scientific公司;Venusil®XBP C18(5 μm,4.6 mm×150 mm,15 mm)分析柱,Agela Technologies公司。流动相0.1%甲酸水溶液(A)- 0.1%甲酸乙腈溶液 ( B ),层析程序为0~4 min 5% B,4~30 min 5%~40% B,30~35 min 40%~80% B,35~45 min 80% B,45~45.1 min 80%B~5% B,45.1~62 min 5% B;总流速 400 nL/min;上样量10 μL。
质谱条件:正离子模式扫描,一级扫描范围50~2 200m/z,分辨率20 000,碎裂模式诱导碰撞解离(CID),碰撞气体Ar,离子源电压1 500 V,毛细管温度150 ℃,二级扫描范围依赖于一级母离子质荷比自动选择。
切下非还原SDS-PAGE凝胶中细胞色素C(2018001批次)样品的2.5×104条带,经消化脱色、还原烷基化、胰蛋白酶酶解处理后,进行LC-MS/MS分析并与猪源蛋白库搜寻鉴定。用质谱采集软件Bruker Daltonics micro TOF control提取质谱数据,利用 Data Analysis 软件标峰后,采用MASCOT search engine version 2.3.01搜索猪源蛋白库,进行鉴定。
2 结果与讨论
2.1 疏水层析图谱
新工艺制备细胞色素C的疏水层析图谱见图1。由图1可知:盐析后含细胞色素C的上清液与Butyl-Sepharose FF层析柱在高盐浓度缓冲液中吸附,低盐浓度洗脱液中解吸附,在约120 min处出现1个极大吸收峰,该峰为细胞色素C峰,其余小吸收峰多为杂蛋白峰。结果表明采用疏水层析法能有效分离细胞色素C与杂蛋白。

图1 细胞色素C疏水层析图谱Fig.1 Hydrophobic chromatography of cytochrome C
2.2 SDS-PAGE分析细胞色素C结果
非还原SDS-PAGE分析细胞色素C的结果见图2。由图2可知:所有细胞色素C供试样品都含有1.22×104的猪心细胞色素C,其中细胞色素C标准品、对照品、2018001批次样品还包含1个大小约2.5×104的蛋白,余下几批次样品不含该蛋白质。通过比较这些样品的生产工艺发现,细胞色素C标准品、对照品、2018001批次样品制备过程中均使用了三氯乙酸作为沉淀剂,而其余7个批次样品制备过程中未使用三氯乙酸。这提示约2.5×104大小的蛋白分子条带与生产过程中使用三氯乙酸沉淀剂之间具有某种关联性,而三氯乙酸沉淀剂容易引起细胞色素C聚合,该蛋白分子量约2.5×104,恰为细胞色素C分子量(1.22×104)的2倍,所以可以推断:2.5×104处的蛋白很可能是2个细胞色素C分子聚合而成的二聚体。
二硫键是蛋白质聚合常见的连接方式,为了验证该蛋白是否为二硫键连接的细胞色素C二聚体,因此采用还原性SDS-PAGE进一步分析该蛋白,若经还原剂处理后2.5×104附近的条带消失,且不出现其他新的条带,则说明该蛋白是由两个细胞色素C分子通过二硫键连接而成的二聚体。具体分析结果见图3。由图3可知:泳道1~3为细胞色素C经还原剂处理后的蛋白样品,泳道4~6为未经还原剂处理的细胞色素C蛋白样品,以上所有样品都显示存在分子量约2.5×104的条带,这说明此2.5×104蛋白不是由肽链间二硫键连接的二聚体,而可能是2个细胞色素C分子以非二硫键方式形成的二聚体,为了验证此猜想需要进一步分析二聚体的结构。其中,经还原剂处理蛋白样品的该条带略高于未经还原剂处理的蛋白样品的对应条带,笔者认为这可能是因为未还原的蛋白质可能不会被SDS完全饱和[16],这使得该类蛋白的分子质量测定结果不如完全变性的蛋白的分子质量测定结果明确,从而造成电泳迁移速度的差异。
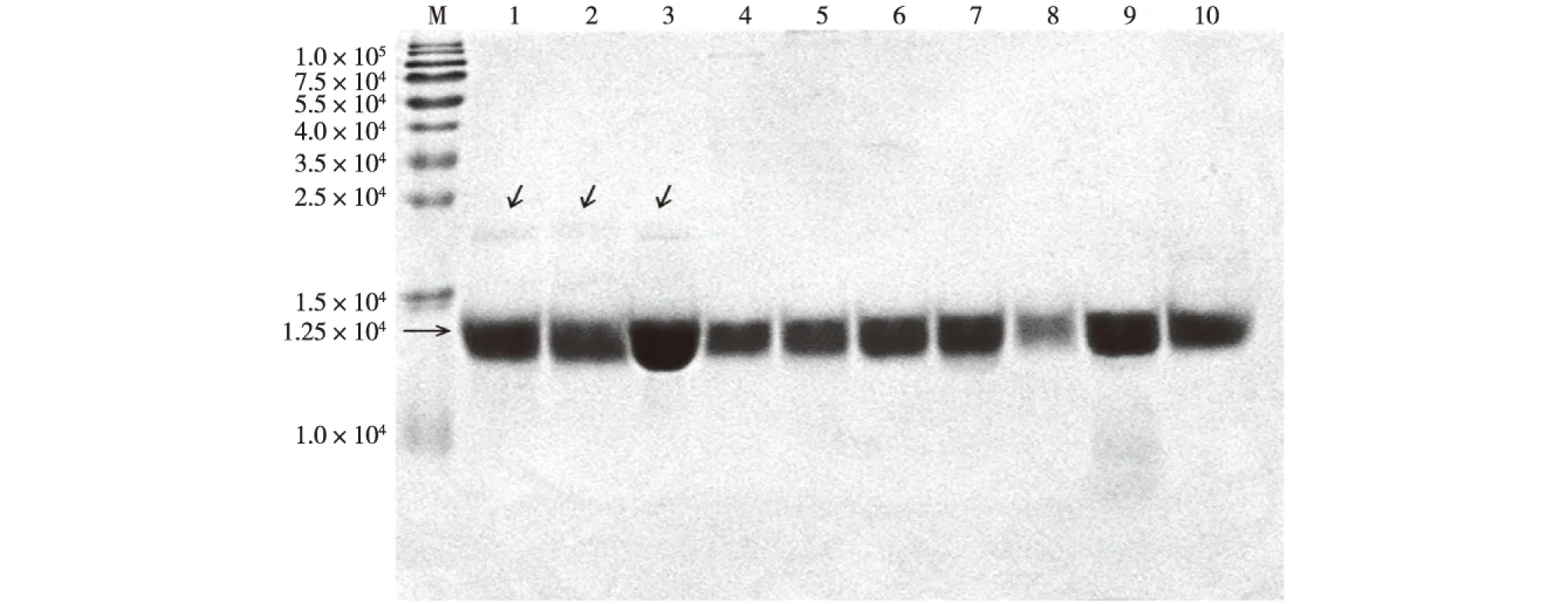
M—标准蛋白;1—标准品; 2—对照品;3—2018001批次;4—2018002批次;5—2018003批次;6—2018004批次;7— 2018005批次;8—2018006批次;9—2018007批次;10—2018008批次图2 细胞色素C非还原SDS-PAGE分析Fig.2 Non-reduced SDS-PAGE analysis of cytochrome C

M—标准蛋白;1—标准品+还原剂;2—对照品+还原剂;3—2018001批次+还原剂;4—非还原标准品;5—非还原对照品;6—非还原2018001批次图3 细胞色素C还原性SDS-PAGE、非还原SDS-PAGE分析Fig.3 Reduced and non-reduced SDS-PAGE analysis of cytochrome C
2.3 LC-MS/MS分析与鉴定细胞色素C二聚体结果
为了验证分子量约2.5×104的蛋白是否为细胞色素C二聚体,需要进一步分析其氨基酸序列,因此对传统工艺制备的细胞色素C样品(2018001批次)非还原SDS-PAGE图谱中的2.5×104蛋白质进行LC-MS/MS分析,图4为该蛋白分子的一级质谱图,肽段信息如表1所示。对多个肽段进行了二级质谱分析,图5列举了其中最长的一段18肽(氨基酸序列为GITWGEETLMEYLENPKK,分子量为2 137.0)的一级质谱图,选择m/z=1 069.5的双电荷正离子为母离子,得到该18肽的二级质谱图,见图6。将分析得到的2.5×104蛋白的质谱数据采用Mascot与猪源蛋白库搜寻鉴定,结果见表2。由表2可知:该蛋白与猪源细胞色素C比对有最高得分3 461,有最大的覆盖率76%,同时结合其他蛋白的分子量分析,可以推测该2.5×104大小的蛋白只含有细胞色素C分子;再结合经还原剂处理的细胞色素C样品的SDS-PAGE电泳结果可知,此蛋白是由2个细胞色素C以非二硫键的其他某种共价键连接而成的二聚体。
细胞色素C二聚体的存在会降低细胞色素C原料药的纯度,增大引发过敏的概率,影响产品稳定性。由于细胞色素C二聚体自身不如细胞色素C稳定,且在细胞色素C原料药中含量极低,又与主成分性质相近,不易检测,须从源头上控制,因此应尽量在细胞色素C制备过程中不使用三氯乙酸,以免产生细胞色素C二聚体。

表1 细胞色素C二聚体的LC-MS/MS分析的肽段信息

图5 荷质比1 069.5的一级质谱图Fig.5 Mass spectrum of ion with mass to charge ratio of 1 069.5

图6 荷质比1 069.5的二级质谱图Fig.6 MS/MS of ion with mass to charge ratio of 1 069.5

序号蛋白序列号蛋白描述分子量得分覆盖率/%1p62895细胞色素C11 8103 461762A0A286ZW70肽基脯氨酰顺反异构酶23 8271 895623A0A287B5W2未标记蛋白26 5631 007234A0A286ZY95纤维连接蛋白1265 75692685A0A287AKA2未标记蛋白34 588903486F1SGG3角蛋白 165 006706117I3LDS3角蛋白1059 230623168F1SLQ7线粒体核糖体循环因子29 099552379P02189肌红蛋白17 0745255110A0A287AEL2角蛋白1456 30044327
3 结论
主要针对细胞色素C生产工艺的重要因素——三氯乙酸对细胞色素C质量的影响开展研究,采用SDS-PAGE分析不同工艺下的细胞色素C样品,发现生产工艺中有使用三氯乙酸沉淀剂的细胞色素C标准品、对照品、传统工艺制备的细胞色素C样品(2018001批次)在约细胞色素C分子量2倍大小处出现明显条带(约2.5×104),而采用无三氯乙酸沉淀剂的生产工艺制备的细胞色素C样品(2018002~2018008批次)没有此条带。根据三氯乙酸会使细胞色素C变性与聚合的性质,推测该蛋白可能是细胞色素C二聚体。
进一步采用LC-MS/MS分析与搜寻鉴定该蛋白,发现传统工艺制备的细胞色素C 样品(2018001批次)中该蛋白与猪源细胞色素C的相似性得分最高(3461),覆盖率最大(达到76%),并结合相关蛋白质的分子量分析,可以证明该蛋白为细胞色素C二聚体。
本研究首次探明了细胞色素C制备过程中三氯乙酸沉淀剂易导致细胞色素C形成二聚体,说明三氯乙酸是导致细胞色素C变性与聚合的重要因素之一,这为细胞色素C工艺优化与产品质量提高提供了理论依据与改进方向。
