肠道菌群与白塞病发病的关联
2019-12-24孙鹿希郑文洁
孙鹿希,郑文洁
白塞病(Behçet’s disease,BD)是一种慢性复发性血管炎性疾病,以口腔、生殖器溃疡,眼炎和皮肤病变为主要特征,并可累及消化道、神经、血管等重要脏器和组织。BD多发生于青壮年,病情反复,有较高的致残率和致死率,严重影响患者的生存质量。研究表明BD是遗传因素及环境因素共同参与致病的疾病[1-2]。BD具有较强的地区特异性和种族差异,高发于中国、日本、中东和地中海地区[3],又称为“丝绸之路病”。BD多数为散发病例,但也有一定家族聚集性,人类白细胞抗原(HLA)-B51是已知与BD发病风险相关最强的遗传易感因素[4]。但 HLA-B51仅增加20%遗传易感风险[5],候选基因和全基因组关联研究(GWAS)揭示了更多BD相关基因参与其发病[6-7],除了遗传因素外,细菌、病毒等微生物因素也与BD发病密切相关。BD患者存在多种免疫失衡表现,例如Th1及Th17细胞过度激活、调节性T细胞(Treg细胞)数量及功能异常。人体微生物,尤其是肠道菌群在调节Th1,Th17和Treg等免疫细胞中起到至关重要的作用。近年来,多项研究显示肠道菌群紊乱与炎症性肠病、1型糖尿病、多发性硬化症和类风湿关节炎等多种自身免疫性疾病发病相关[8-11]。本综述旨在讨论肠道菌群组成与BD的关联以及它在该疾病发展中的潜在作用。
1 肠道菌群特点和分析方法
人体内存在大量的共生微生物,特别是肠道微生物,对维持人体内环境稳态起着非常重要的作用。其中,肠道微生物由肠道内的细菌、真菌和病毒等各种微生物构成,是人体最庞大的微生态系统,已知种类超过1000种,其编码的基因含量远远超过人类基因组,又被称为人体的“第二基因组”。
目前已知的肠道细菌超过50个门,主要包括厚壁菌门(Firmicutes)、拟杆菌门(Bacteroidetes)、变形杆菌门(Proteobacteria)、放线菌门(Actinobacteria)和梭杆菌门(Fusobacteria)。按照功能可分为三大类,包括(1)与人体共生的生理性细菌,如双歧杆菌和乳酸菌等,可以通过能量代谢、发挥免疫刺激等功能维持微生物环境稳态;(2)条件致病菌:如肠杆菌、肠球菌和链球菌等,在机体免疫监视低下时可成为致病菌;(3)致病菌:如变形杆菌、假单胞菌、金黄色葡萄球菌和梭杆菌等,可通过菌群易位、产生毒素和对机体有害的物质促进疾病发生。
传统微生物研究采取单一菌种培养的方法进行,但超过99%的微生物无法在体外进行培养,极大的限制了人们对微生物的认知。随着高通量测序和宏基因组关联分析等技术在人类疾病研究中的广泛应用,人类对肠道微生物多样性有了更深入的了解。其中,基于16s rRNA的分子测序技术通过检测细菌16s rRNA的全序列或V3、V6、V8可变区,达到对菌群结构的定性和相对定量分析,已被广泛应用于肠道等人体微生物研究中。但16s rRNA测序具有操作技术环节多、影响因素复杂、难以标化等不足。宏基因组学(Metagenomics)是通过以全部微生物群体基因组(又称为宏基因组)为研究对象,对样本的总DNA直接进行高通量测序,然后进行序列组装和基因注释,可以获得完整的微生物基因组信息,从而发现肠道菌群结构改变、系统全面地研究微生物与其生存环境间的关系。
越来越多的证据显示,肠道菌群具有多种功能,除了物质代谢、黏膜屏障、免疫调控及宿主防御等,还能合成多种小分子代谢产物,在疾病的发生发展中起着着重要的作用。
2 BD患者的肠道菌群改变
Consolandi等[12]的研究首次揭示了BD患者与健康对照之间存在的肠道菌群差异及潜在机制。该研究收集了地中海地区的22例BD患者及16名与其共同生活的健康亲属的肠道菌群,通过16s rRNA测序方法比较发现,两者的肠道菌群在门级别分布大致相同,但BD患者的肠道菌群的种属多样性显著降低,尤其是罗斯氏菌(Roseburia)和罕见小球菌(Subdoligranulum)两种重要梭菌屬的丰度显著减少。
短链脂肪酸(short chain fatty acid,SCFA)是由肠道菌群产生的参与宿主免疫调控的重要代谢产物,主要包括乙酸、丙酸和丁酸[13]。大量研究表明SCFA具有诱导细胞分化[14]、凋亡[15]和抗炎[16-18]等重要生理功能。目前已知短链脂肪酸是重要的组蛋白去乙酰化酶(histone deacetylases, HDAC)抑制剂,特别是丁酸被证实具有广泛HDAC抑制作用。SCFA可以通过抑制外周血单个核细胞[17-18]、巨噬细胞[16]和树突细胞[19-20]的HDAC抑制炎症反应。体外实验显示,SCFA可以通过抑制HDAC使NF-κB失活并下调TNF-α的表达[17-18],从而抑制局部促炎细胞因子产生。除了影响固有免疫细胞,丁酸等SCFA还可以通过其HDAC抑制作用,促进Treg细胞分化和免疫抑制功能增强,从而维持结肠的免疫稳态[20-22]。
该研究通过检测粪便的短链脂肪酸水平,揭示了BD患者肠道菌群产生的丁酸含量显著下降[12]。而丁酸盐水平降低可通过上述机制促进各种炎症成分的表达、Th17/Treg等细胞分化和功能异常,从而导致宿主免疫稳态失衡。同时BD患者的肠道罗斯氏菌与丁酸水平存在显著正相关性。提示肠道菌群中梭菌属等产短链脂肪酸菌减少所导致的短链脂肪酸生成不足可能与BD的发生相关。
日本的一项研究指出,BD患者的11种肠道细菌亚群的相对丰度与健康对照存在差异[23]。该研究通过16s rRNA测序对比了12例BD患者和12名健康对照的肠道菌群,结果显示BD患者中双歧杆菌(Bifidobacterium)和爱格士菌(Eggerthella)两种菌属显着增加,普雷沃菌(Prevotella)和巨单胞菌属(Megamonas)两种菌丰度减少。有研究指出口服双歧杆菌可以诱导出无菌小鼠的Th17细胞激活和关节炎表现[24-25]。双歧杆菌与肠道内乳酸的产生和pH调节密切相关[26],部分梭菌属可以利用乳酸产生丁酸盐或丙酸盐等短链脂肪酸[27-28],而且梭菌属与调节性T细胞的活化相关[22]。BD患者中的双歧杆菌和梭状杆菌的失衡可能引起局部酸碱平衡和黏膜免疫系统紊乱。与Consolandi等[12]研究不同的是,该研究中个体和群体的肠道菌群多样性与正常人无明显区别,菌群的丰度变化也与疾病活动度无明确相关性。这可能与该研究所招募的BD患者病情活动度相对较低相关。
以上两项研究主要基于16s rRNA测序分析,该测序方法有一定的局限性,例如由于扩增子偏倚造成的微生物组成偏差以及无法达到菌种和菌株水平的鉴定。为了更全面的探究BD患者微生物的组成,Ye等[29]首次采用宏基因组的测序技术研究BD患者和健康人肠道菌群组成的差异。结果显示两者的主要菌群分布一致,主要为拟杆菌门、厚壁菌门和变形菌门,但在96个菌属和23个菌种的丰度中存在显著差异。BD患者中富集的菌属主要包括子囊菌真菌、变形杆菌和放线菌。同时,BD患者中富集的菌种主要包括多种窄食单胞菌属、放线菌属和棒状杆菌中的条件致病菌。
通过LEfSe分析和差异基因聚类分析显示,硫酸盐还原细菌(Bilophilaspp.)和副杆状菌属(Parabacteroides)等多种条件致病菌在BD患者中显著富集,而产甲烷菌(Methanoculleusspp.和Methanomethylophilusspp.)以及产丁酸盐细菌(Clostridiumspp.)在健康人群中富集。同时基于宏基因组物种分析谱(metagenomic species,MGSs)确立了13个差异MGSs,从而构建了能够区分BD患者与健康者的疾病监测模型,经分析该模型AUC=81.1%(95%CI=70%~92%),为通过肠道菌群检测识别BD患者提供依据。
3 BD患者的口腔菌群改变
人口腔微生物是人体共生菌群的重要组成部分,口腔内存在700多种丰富多样的菌群[30]。其中,链球菌属(Streptococcus)、韦荣菌属(Veillonella)以及普氏菌属(Prevotella),是占最大比例的微生物菌属。近年来许多研究显示,除了肠道菌群,口腔菌群与全身疾病的发生也存在密切关联,包括炎性肠病、胰腺癌和类风湿关节炎等[9,31-32]。而且由于唾液样本易于获取,口腔微生物有可能成为许多疾病的便捷使用的生物标志物来源。
BD患者常具有特征性的反复发作的口腔溃疡,既往研究显示BD和牙周病存在相关性,BD患者通常具有比其他类型口腔溃疡患者更差且与BD疾病严重程度相关的牙周评分[33],均提示BD患者口腔微生物与免疫反应之间的相互作用可能在疾病的发生中起到重要作用。
英国的一项研究首次探索了BD患者口腔微生物组成[34]。应用DNA微阵列分析方法,检测了BD与复发口腔溃疡(recurrent aphthous stomatitis,RAS)患者及健康对照的唾液和口腔黏膜微生物成分。结果显示,BD患者存在口腔健康受损,BD患者龋齿指数、牙龈指数、菌斑指数和牙周探诊深度和龈沟出血指数均较正常人高,且与RAS患者无差别。和RAS患者相比,BD患者非溃疡部位龋齿罗菌(Rothiadenticariosa)定植增加,而溃疡处唾液链球菌(Streptococcussalivarius)明显增加。与健康对照相比,BD患者犬血链球菌(Streptococcussanguinis)明显增加。既往研究曾多次报道以犬血链球菌为代表的链球菌属感染与BD之间存在关联。链球菌属的细菌和单纯疱疹病毒-1与热休克蛋白等人体具有的蛋白质具有高度同源性,可引起BD遗传易感个体的交叉反应[35]。此外,近年也有研究证实在BD患者口腔中存在一些非典型链球菌的增多,如口腔犬血链球菌的特定菌株可以促进免疫细胞分泌IL-6、IL-8和肿瘤坏死因子(TNF)-α等促炎因子[33,36]。虽然暂无明确直接证据指出BD患者口腔微生物失衡是致病性还是反应性,但这些证据提示在遗传易感的基础上,口腔等细菌感染引发的免疫反应可能促进BD进展。同时,通过患者特异性、个体化的益生菌治疗恢复溃疡部位的口腔微生物群落的平衡可以作为未来口腔溃疡治疗的新方向。
另一项来自美国密歇根大学的研究应用了16s rRNA测序方法检测了BD患者唾液中微生物的组成。该研究同样指出BD患者的口腔微生物组与对照组相比存在显著差异,主要表现为菌群多样性的减低。在BD患者中,副流感嗜血杆菌(Haemophilusparainfluenzae)和拟普雷沃菌属(Alloprevotella)的丰度显著增加。两种菌均是革兰氏阴性杆菌,且与牙龈炎症相关[37-38]。该研究还探索了免疫抑制剂的应用以及HLA-B基因型与口腔细菌群落组成间的关系。结果显示,在BD患者中,是否服用免疫抑制剂以及是否具有高风险等位基因与口腔菌群的相对丰度之间没有明确关联。此外,既往研究显示,牙周治疗可以改善口腔环境,但唾液微生物组成在治疗前后无明显改变[39]。与此一致的是,BD患者经过牙科治疗后口腔环境改善,牙菌斑微生物组趋于正常,但唾液微生物组未见明显改变,提示唾液微生物成分更稳定,可作为潜在的BD生物标记物。
4 肠道及口腔微生物改变在BD发病中的潜在作用机制
由于口腔微生物可以通过唾液从口腔进入下游的消化道,其与肠道菌群也存在着值得探索的相关性。研究显示,将从炎症性肠病患者的唾液当中获取的克雷伯氏菌,直接接种到无菌小鼠的口腔,结果发现这种菌可以通过吞咽作用,加重炎症性肠病的反应[40]。Ye等[29]发现BD患者唾液中存在双歧杆菌、普雷沃菌和斯卡多维亚菌属(Scardovia)的富集,且BD患者的口腔和肠道中均检测到普雷沃氏菌的异常,也对口腔和肠道菌群间存在关联并共同参与致病有所提示。
Ye等[29]还进一步分析了肠道菌群功能改变。京都基因与基因组百科全书(Kyoto encyclopedia of genes and genomes,KEGG)是整合了基因组、化学和系统功能信息的数据库,能够全面提供如代谢、膜转运和信号传递通路等信息,被广泛应用与菌群功能分析中。其中同源KEGG(KEGG Orthology,KO)是基于同源基因具有相似功能的假设,把和每个功能已知的基因同源的基因都归为一类,用该基因的功能作为这个KO的功能,以此用不同物种的基因功能研究进行相互扩充,建立全面的基因功能数据库。KEGG模块是人为定义的功能单元的集合,被用于已测序基因组的注释。BD患者中与氧化还原过程相关的KO有所增加,而能够降解细胞壁中的肽聚糖而具有消炎作用的溶菌酶相关的KO则显示减低。在KEGG模块水平,BD患者中的荚膜多糖运输系统、types Ⅲ和types Ⅳ分泌系统显著增强。BD患者常合并葡萄膜炎,该研究利用粪菌移植的技术将BD患者的粪便菌群移植到葡萄膜炎小鼠动物模型中。组织学分析显示BD受体组小鼠的视网膜结构严重紊乱,存在大量炎症细胞浸润和IL-17和IFN-γ等炎症因子分泌增加。该研究首次应用动物模型揭示了BD患者的粪便可诱导或加重葡萄膜炎。
基于以上结果,我们总结肠道菌群紊乱参与BD的发病可能存在下述机制(图1)。近年研究中均发现BD患者的肠道菌群中有多种致病菌和条件致病菌富集,而可以产生短链脂肪酸和产生甲烷的有益菌群则相对减少。其中,BD患者肠道硫酸盐还原菌的富集可能导致具有促炎作用的硫化氢的过度产生,硫化氢与肠道的健康息息相关,它可以作为蛋白质酵解产生的毒素直接损害肠腔上皮或血管内皮细胞,损害其屏障功能,也可转化为硫代硫酸钠,在炎症期间可进一步氧化成连四硫酸盐,促进沙门菌及部分变形菌类致病菌的生长。条件致病菌在BD患者肠道中的富集可引起其效应分子和病原体相关模式分子(pathogen-associated molecular pattern,PAMP)侵入肠上皮细胞或血管内皮细胞,被模式识别受体TLR2/TLR4识别,诱导Th1和Th17过度活化,从而导致导致肠道和血管炎症,并最终导致BD的发生。
另一方面,产丁酸菌和产甲烷菌水平降低可能导致炎症途径的抑制作用下降,从而诱发BD全身性疾病表现。例如,产甲烷菌产生的甲烷可以改善氧化应激损伤并且可以抑制各种组织和器官中的炎症反应[41-43]。产丁酸菌可以发酵膳食纤维产生丁酸盐[44],后者的减少可导致肠上皮屏障功能障碍[27],引起Treg细胞数量减少,而Th17等致病性免疫细胞活化及其分泌的致炎因子增多,这与BD患者中发现的免疫细胞失衡表现相一致[45-47]。
5 总结
综上所述,BD患者具有特征性的肠道微生物和口腔微生物组成改变(表1),黏膜微生物群结构的变化可通过影响黏膜屏障功能、触发天然免疫、诱导Th1和Th17过度活化、Treg数量减少等多种机制促进BD发病(图1)。进一步干预性研究,如饮食干预、益生菌治疗、粪便微生物移植等重建BD患者菌群的组成可能对疾病的防治提供新方法。
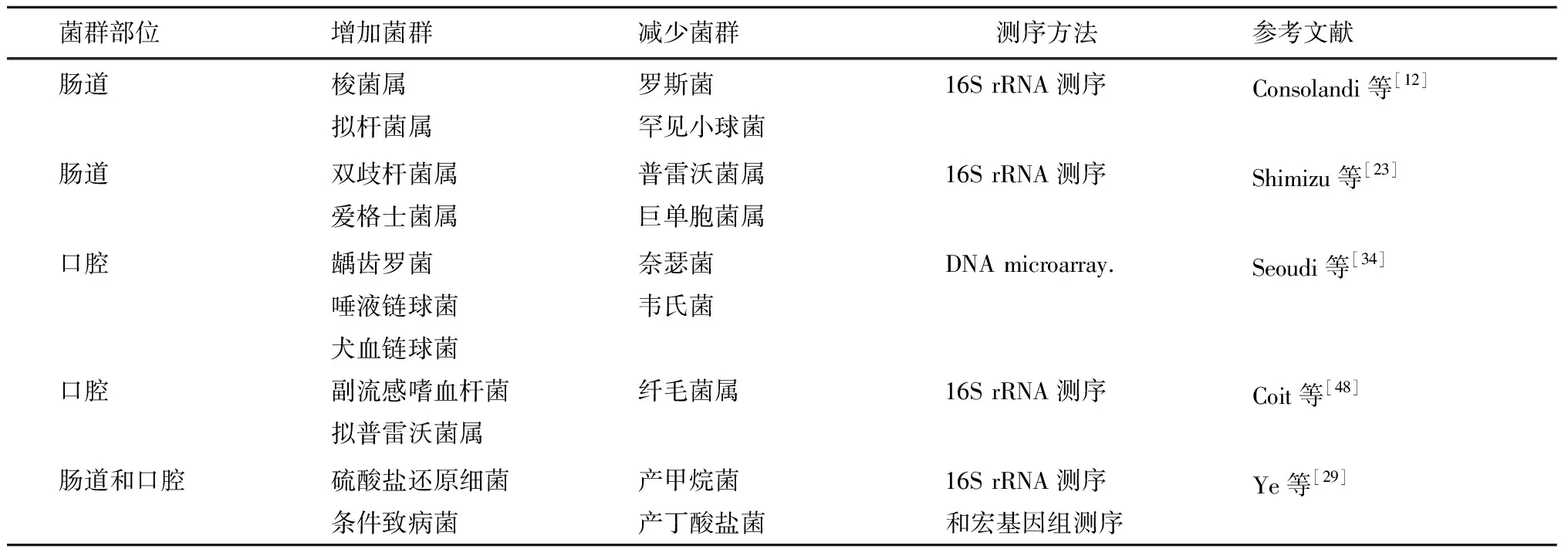
表1 BD患者肠道和口腔菌群组成改变Table 1 Altered composition of gut and oral microbiota in BD patients
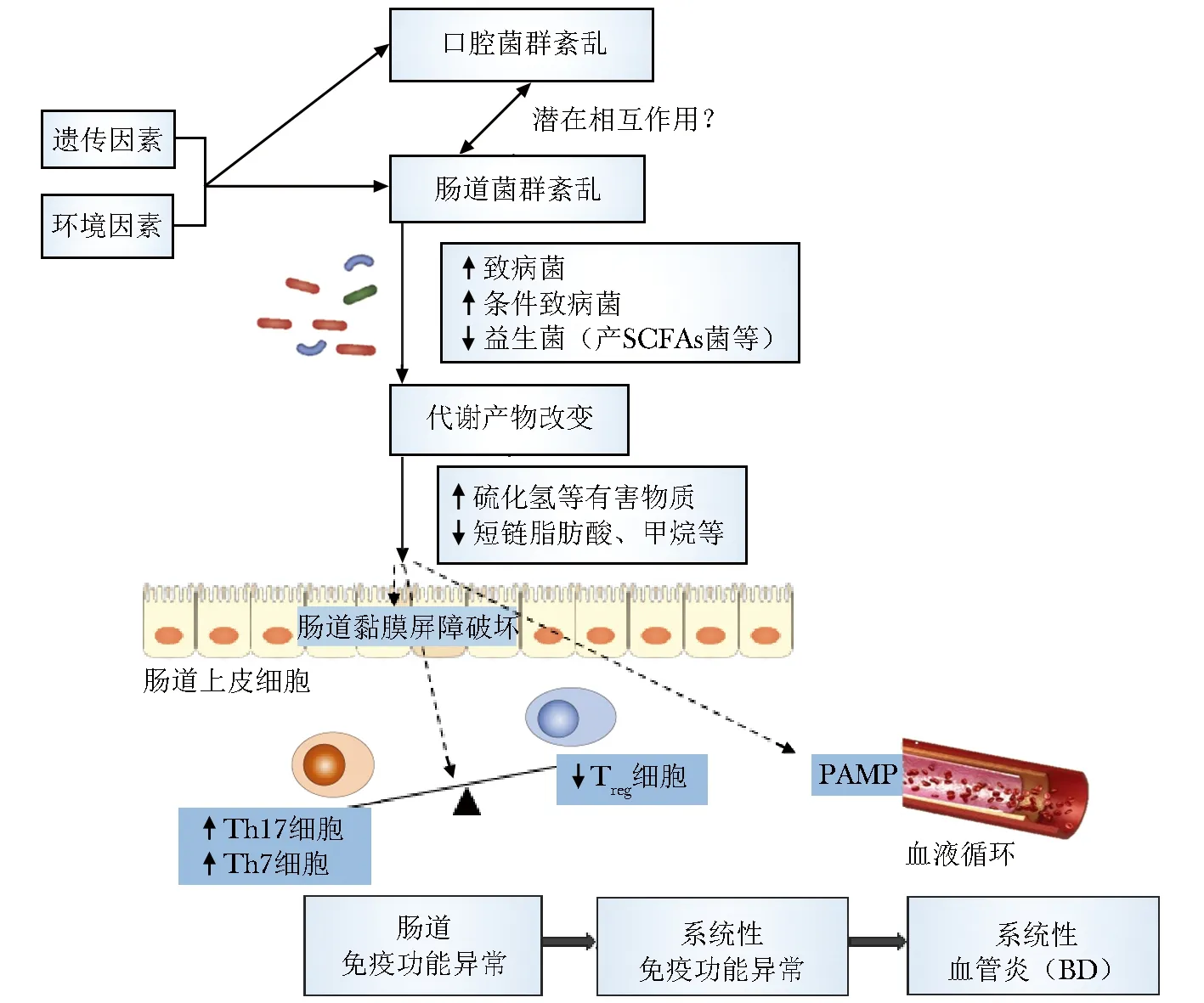
图 1肠道菌群紊乱促进BD发病的潜在机制
Fig1Potential mechanism of dysbiosis of gut microbiota in the pathogenesis of BD
在遗传和环境因素影响下,BD患者出现肠道菌群紊乱,表现为多种致病菌和条件致病菌富集,而产SCFAs等有益菌群则相对减少。致病菌生成多种促炎作用的有害物质(如硫化氢等),而益生菌生成的短链脂肪酸等物质减少;菌群的代谢产物异常可破坏肠腔上皮或血管内皮细胞,引起屏障功能障碍,并引起其效应分子和病原体相关模式分子(PAMP)侵入肠上皮细胞或血管内皮细胞,被模式识别受体识别,直接或间接诱导Th1和Th17过度活化、Treg细胞数量下降,从而导致导致肠道和血管炎症,并最终导致BD的发生
