炎症通路在强直性脊柱炎发病中作用的转录组学研究
2019-12-20李辰璐方锦霞罗京朱小春王晓冰
李辰璐,方锦霞,罗京,朱小春,王晓冰
(温州医科大学附属第一医院 风湿免疫科,浙江 温州 325015)
强直性脊柱炎(ankylosing spondylitis,AS)是一种主要累及脊柱、骶髂关节等中轴关节和膝关节、髋关节等外周关节的慢性炎症性疾病。其主要的病理改变是附着点炎,严重者会出现骨侵蚀和关节破坏,甚至关节融合强直,对患者的生活带来严重的危害。在全球范围,AS的发病率为1.0%~1.4%[1],与遗传背景密切相关。
AS的易感等位基因人类白细胞抗原B27(human leukocyte antigen B27,HLA-B27)的阳性率分布与全球的发病率分布相一致[2],它是迄今为止已知的复杂疾病中相关性最强的一个等位基因[3]。此外,全基因组研究发现某些非HLA易感基因在AS的发展中起重要作用[4]。转录组是由基因组转录的一系列RNA分子组成,包括mRNA、tRNA、rRNA和其他的调节非编码RNAs。与基因组不同,转录组是动态的,伴随着个体的生命进程不停地变化进展,而且还会针对环境刺激和内部生物信号做出快速的反应。这些基因的差异表达和调节是真核生物细胞分化的基础,扩展了静态的基因组的视野,使机体内不同类型的细胞能表达出完全不同的功能[5]。在人类疾病的研究中,比较基因表达的分析为疾病的遗传学研究提供了的重要信息。通过上调或者下调它们的表达成分即能决定有无患病群体的表型差异。
对AS患者血标本的转录组分析发现炎症作用基因以及调节软骨和骨代谢的候选基因[6]。来自Gene Expression Omnibus数据库的分析证明了AS患者较对照者高表达的差异基因主要富集在抗原加工和抗原提呈,细胞因子受体相互作用、自然杀伤细胞介导的细胞毒性等通路[7-8]。根据现有研究,除ERAP1、IL23R和RUNX3的少数基因确认与AS发病过程相关,还需要更进一步的研究来确认这些通路在疾病过程中的参与以及发掘更多相关基因[9]。然而,不同地域和人种的遗传背景具有多样性[10]。AS之前的转录组研究大多针对非汉族人群。本研究旨在通过探讨汉族人群AS患者与正常人转录组差异,分析AS发病机制,为治疗AS寻找新的靶点。
1 资料和方法
1.1 一般资料 选取温州医科大学附属第一医院2017年住院的53例AS患者作为病例组,其中男46例,女7例。同期健康体检者53例为对照组,男46例,女7例。病例组纳入标准:18~70岁且有>3个月的炎性腰背痛,符合1984年纽约分类标准[11]。排除标准:合并其他免疫系统疾病,如系统性红斑狼疮、原发性干燥综合征、类风湿性关节炎、银屑病性关节炎、炎症性肠病性关节炎,合并重要脏器功能不全,合并慢性感染性疾病(包括乙型病毒性肝炎、结核)、肿瘤、心脑血管疾病、精神障碍等。本研究经医院伦理委员会批准,所有受检者签署知情同意书。每位受检者抽取外周血5 mL,加入PBS与淋巴细胞分离液后离心取白膜层,洗涤获得外周血单核细胞(peripheral blood monocytes,PBMCs)。将从PBMCs样品中提取的RNA反转录为cDNA,并在 Illumina平台上测序。
1.2 差异表达基因(differentially expressed gene,DEGs)筛选 使用DEseq2(18)软件包对RNA-seq数据进行标准化,并进行二维主成分分析(principal component analysis,PCA)和层次聚 类,以显示病例组和对照组之间的相似性和差异。使用DEseq2进行差异基因表达分析,如果基因符合以下标准,则该基因被认为是DEGs:DEseq2中的错误发现率(false discovery rate,FDR)<0.05和log2倍变化(FC)<-1或log2 FC>1。使用ggplot2软件包在R软件中构建可视化不同对象之间所有DEG的火山图,并使用R软件包pheatmap绘制DEG的热图。
1.3 差异表达基因的GO(gene ontology)、KEGG(Kyoto Encyclopedia of Genes and Genomes)富集分析 采用ClueGo插件[12]在Cystoscape[13]中整合了GO术语,并分别创建了具有上调基因的生物过程 网络。采用Bonferroni法进行校正,P阈值为0.05。信号通路分析在Kobas3.0[14]的在线网站(http://kobas.cbi.pku.edu.cn)上进行,包括KEGG途径、Reactome、BioCyc和PANTHER信号通路分析,并使用GOplot绘图。
1.4 蛋白相互作用网络分析(protein-protein interaction,PPI) DEGs的PPI数据是从用于检索相互作用基因的搜索工具(STRING)版本11.0(https://www.string-db.org/)[15]下载的数据。然后,通过Cytoscape软件建立并可视化PPI网络,并使用插件MCODE[16]确定DEG的相互关联程度。
1.5 细胞因子分析 本试验另外入选了30例AS患者及30位健康对照。纳入及排除标准同前。所有受检者签署知情同意书后抽取外周血5 mL,提取血浆。ELISA法测定患者外周血浆中IL-2、IL-4、IL-6、IL-10、TNF-α和IFN-γ等细胞因子水平。
1.6 统计学处理方法 采用Rv3.6.1 for Linux软件DEseq2、ggpubr统计软件包进行统计学分析。差异基因分析使用Wald检验。2组间细胞因子比较使用秩和检验。P<0.05为差异有统计学意义。
2 结果
2.1 AS患者与对照组的基因表达模式差异 使用DEseq2对比AS研究组与对照组基因表达差异,基于标准(P<0.05 and |logFC|>1),找到153个DEGs,其中149个基因上调,4个基因下调(见图1)。表明除HLA-B27以外,还有一系列的基因参与了AS的发病。其中部分基因在既往的文献报道中提及与AS相关,如THBD[17]、IL1B[18]、CXCL8[19]、CCL7[20]等。提取基因表达值,绘制层次聚类热图以显示所有的差异DEGs,可以看到2组的基因表达谱存在明显的差异,见图2。
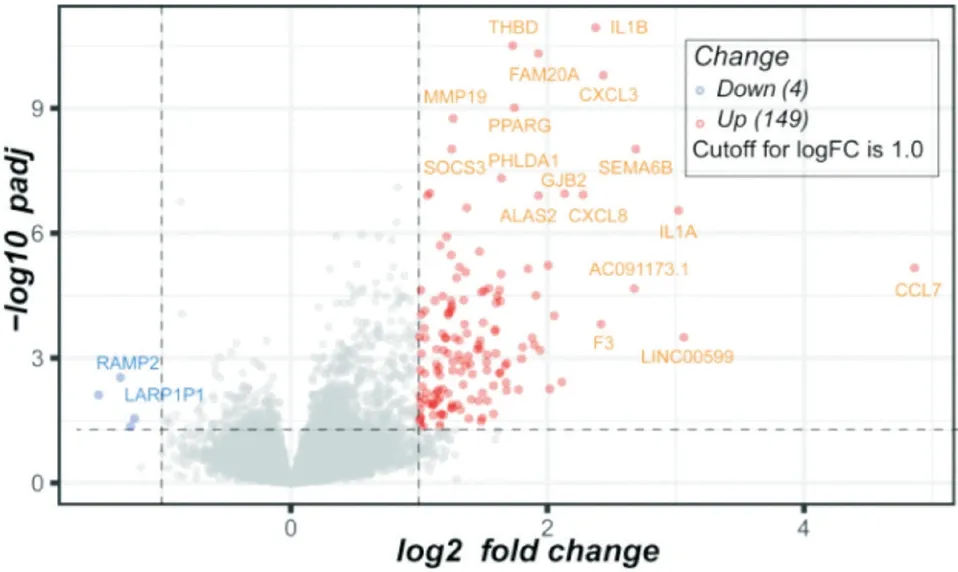
图1 AS中所有DEGs火山图

图2 AS组和对照组DEGs热图
2.2 AS组与对照组的DEGs涉及的信号通路 对发现的DEGs进行KEGG分析,发现了几条重要的信号通路,包括细胞因子间相互作用、TNF信号通路A/1(视紫红质样受体)、GPCR配体结合、趋化因子受体结合、CCKR信号图、血液凝固、趋化因子介导的炎症和细胞因子信号通路等,见图3。

图3 DEGs通路图
2.3 AS组与对照组的DEGs编码蛋白的相互关系 对涉及KEGG途径的DEGs进行了PPI分析。在由STRING数据库构建的PPI网络中,大多数基因,包括CXCL8、IL1B、CXCL1、CCL2、PTGS2、EDN1 和 SERPINE1,与KEGG途径中涉及的其他DEGs相互作用(见图4),表明这些基因可能通过相互作用参与AS的发病。

图4 通过STRING完成的DEGs的PPI图
2.4 AS组与对照组的DEGs的生物功能注释 DEGs在生物学过程(BP)中显著富集,包括粒细胞迁移(GO:0097530),中性粒细胞迁移(GO:1990266),发热过程(GO:0001660)等,这些都起着重要的作用。此外,DEGs在分子功能组(MF)中显著富集趋化因子受体结合(GO:0042379)、趋化因子活性(GO:0008009)和过氧化物酶活性(GO:0004601)。总之,GO分析的结果表明,大多数DEGs在粒细胞迁移、中性粒细胞迁移、发热过程、趋化因子活性等相关,所有这些都可能与炎症反应有关。见图5。
2.5 炎性细胞因子差异 为进一步验证这些炎症相关基因对AS的作用,对外周血的重要炎症因子进行验证,发现AS组较对照组在IL-2、IL-6、IL-10、TNF-α、IFN-γ等外周血炎性细胞因子水平上升(P<0.05),IL-4因子水平差异无统计学意义(P>0.05)。见图6。
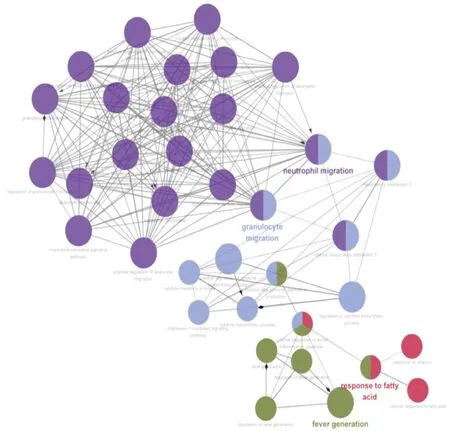
图5 DEGs的ClueGo网络分析图

图6 2组细胞因子相对表达量(每组n=30)
3 讨论
RNA是基因和蛋白之间的关键链接,所以转录组研究已经广泛应用于结缔组织疾病的发病机制及治疗靶点研究。本项目通过高通量的RNA测序技术,发现AS患者和健康对照者存在153个DEGs,其中149个基因上调。而这些基因相互之间密切联系,相互作用,共同参与疾病的发生。生物功能分析提示这些差异基因主要聚集在TNF信号通路、趋化因子信号传导等炎症相关通路。而这个结果又进一步在外周血中的炎性细胞因子的表达水平得到验证。
已有研究发现,破骨细胞分化、INF信号传导途径和与这2种信号传导途径相关的基因,特别是 FCGR2B、STAT2、SOCS3、IFIT1 和IFIT3,可能在AS的发病中发挥作用[21]。本研究发现了大量与TNF信号通路相关的基因,如CXCL3、IL1B、CCL20、CXCL2、 CXCL1、SOCS3、PTGS2 等。已有研究证实TNF信号通路在AS发病过程中的重要作用,而对TNF-α的特异性抑制剂,也已在临床上大量使用于AS患者的治疗,并取得良好的疗效[22]。因此,本研究也为中国汉族人群应用该生物制剂提供了一定的理论依据。
本研究发现在AS患者的外周血细胞中趋化因子/趋化因子受体家族的炎症因子表达显著上调,包括CCL7、CXCL3、CXCL8、CCL2、CCL20、CXCL2、CXCL1。我们对DEGs进行通路分析也发现这些差异表达的基因集中在趋化因子介导的炎症及趋化因子与趋化因子受体结合等通路。生物学功能分析也提示这些基因在粒细胞迁移中的作用。既往研究已经发现趋化因子的定向趋化作用控制着各种免疫细胞到达感染、创伤和异常增殖部位,从而发挥特定的免疫功能[23]。所有这些都支持AS疾病过程中免疫炎症的过度活化,也进一步支持AS是一种兼具有炎症和免疫特色的系统性疾病。
临床病例的外周血检测发现TNF-α炎症相关的关键细胞因子在AS中表达明显上调,进一步验证了测序结果。文献报道IL-2、IL-6、TNF-α[24-25]等细胞因子在AS疾病的病理过程中发挥作用,跟我们的研究结果相符。因此,这些细胞因子也成为AS治疗的靶点,也为细胞因子的生物制剂治疗AS提供理论依据。
本研究的不足是进行RNA测序的样本数相对较少,尚未考虑患者的个体差异和疾病的特质性,仍需要大样本的研究以更加全面可靠地证实本研究的结果。另外,本研究中发现的许多可能参与AS疾病过程的DEGs,尚无法确定其具体作用机制,仍需要进一步通过细胞和(或)动物模型进一步探索。
总之,本研究通过对AS和正常人转录组的研究,发现了炎症相关基因在AS患者中明显表达上调,提示这些基因可能参与AS的发病过程。这些基因主要涉及到TNF信号通路、趋化因子/趋化因子受体等炎症相关的重要组成成分和通路。这些信号通路及炎症因子可成为未来AS诊断标志物和治疗靶点。
