敲除PLAC8蛋白对人胚肾细胞增殖的影响
2019-12-19秦绪慧马立群欧阳聪王海霞浦易之
秦绪慧, 马立群, 欧阳聪, 王海霞, 浦易之, 薛 璐
(中南民族大学 生命科学学院 医学生物研究所 武陵山区特色资源植物种质保护与利用湖北省重点实验室, 武汉 430074)
胎盘特异蛋白8 (Placenta special gene 8,PLAC8)又称onzin或C15,是一种高度保守并富含半胱氨酸,有着独特结构并在组织中特异性表达的蛋白。PLAC8首次发现是在人树突状细胞中,定位于染色体的4q13-4q21区域。PLAC8含有5个外显子,mRNA全长829 bp,编码115个氨基酸,蛋白分子质量为12.4 ku[1-2]。广泛分布于真核生物中的PLAC8不含信号肽,但是在一些哺乳动物中有类似限制酶位点的信号肽。PLAC8在不同物种中氨基酸序列差异较大,说明在进化早期,PLAC8的进化并不稳定[1,3],也提示了PLAC8在不同物种中可能承担着不同的功能角色。
PLAC8的表达调控涉及多种肿瘤的发生与发展,与细胞的凋亡、增殖及分化等生理过程都有着十分密切的关系[1-2]。在细胞分化方面,有研究发现PLAC8是棕色脂肪细胞分化所必需的蛋白,如果PLAC8蛋白缺失可能会导致寒冷耐受不良症、迟发性肥胖和棕色脂肪细胞功能受损[4]。Jimenez-Preitner等发现,在小鼠体内,PLAC8在棕色脂肪组织和白色脂肪组织中均有表达,并且发现PLAC8敲除小鼠会产生迟发性肥胖,棕色脂肪分化异常,产热能力下降[5]。除此之外PLAC8也广泛存在于免疫细胞之中,尤其是巨噬细胞与中性粒细胞。如果在成体上敲除该基因,虽然不会影响吞噬细胞的吞噬作用,但是其吞噬效率会降低,这提示我们PLAC8可能在免疫过程中发挥着十分重要的作用[2,6]。Ledford等发现,PLAC8基因敲除小鼠虽然发育的正常,但是在噬中性粒细胞的功能上表现出明显的缺陷[7]。在调控细胞增殖方面,Zou[8]和Uehara[9]等用人胰腺细胞系体外实验证明,通过siRNA敲除PLAC8蛋白的表达能抑制胰腺癌细胞的增殖。在结肠癌的相关研究中,Li等在结肠癌细胞系中敲除内源PLAC8后发现肿瘤的体积减小,这一过程可能是通过改变ERK2的表达来实现的[10]。与之相反的是,Zou等通过细胞功能实验研究证明,在肝癌细胞中,PLAC8下调会促进细胞存活,促使细胞增殖进而促进肿瘤的形成[9]。由此可见,PLAC8蛋白在不同肿瘤来源细胞系中对增殖具有不同的调控模式,值得进一步研究。
人胚胎肾细胞极少表达细胞外配体所需的内生受体,且比较容易转染,是一个十分常用的表达研究外源基因的工程细胞株。因此在本课题中,我们利用流式细胞术与CRISPR/Cas9基因编辑技术相结合的方法在293T细胞系上进行PLAC8蛋白敲除的研究,最终获得了PLAC8蛋白敲除人胚肾细胞系。随后的一系列功能实验表明,敲除细胞系的增殖相比野生型有明显的下降。进一步地,我们发现PLAC8蛋白的表达下降同时伴随着AKT,ERK2,RAF-1,C-MYC等基因的表达水平改变,提示在293T细胞中,PLAC8可能通过ERK/MAPK信号通路来调控细胞增殖。
1 材料与方法
1.1 材料和试剂
人胚肾细胞株 (293T)购于武汉大学细胞库,PX458质粒、感受态细菌Stbl3、10×T4 Ligation Buffer、T4 连接酶购于武汉淼灵生物,T4 PNK、10×TaqBuffer、dNTP、rTaq酶购于TaKaRa,Fast Digest BpiI、Fast AP、10×Fast Digest Buffer、Protein Ladder购于Thermo Scientific,DTT购于Biosharp,Endo-free Plasmid Mini Kit购于Omega,AxyPrep DNA Gel Extraction Kit购于AxyGen,PBS购于HyClone,血清购于ScienCell,双抗、胰酶、Lipofectamine 3000试剂购于Invitrogen, BCA蛋白浓度测定试剂盒、PMSF、SDS-PAGE凝胶快速配制试剂盒、Tween-20、Beyo ECL Plus、上样缓冲液购于碧云天公司,甲醇 (分析纯)、氯化钠、甘氨酸、磷酸二氢钾、十二水合磷酸氢二钠、氯化钙购于国药集团化学试剂有限公司,Anti-PLAC8抗体购于Cell Signaling Technology,Anti-β-Actin、HRP-goat anti mouse IgG、HRP-goat anti rabbit IgG购于康为世纪。引物合成、基因测序交由武汉擎科生物公司完成。
1.2 sgRNA寡核苷酸链序列及检测引物的设计
首先使用CRISPR在线sgRNA设计网站 (http://crispr.mit.edu/),分别对PLAC8基因5个外显子上的sgRNA的潜在靶点进行预测。根据评分系统,选择得分较高的靶点序列,分别在PLAC8的外显子2和外显子3上设计2个sgRNA (S1及S2),见图1。需要指出的是,若sgRNA的序列第一个碱基不是G,则需要在前面补一个G以取得更好的编辑效率。以该sgRNA序列为模板,设计其互补链,然后分别在sgRNA5′ 端添加CACCG,互补链5′ 端添加AAAC,3′ 端添加C,以此人工合成BpiI酶的酶切位点。sgRNA序列见表1。
为确定细胞是否被编辑,需要对基因组进行测序检测。因此我们使用软件Primer Premier 5分别在两个sgRNA潜在靶点的上下游200~300 bp处各设计了一对检测引物 (PLAC8-AF/AR,PLAC8-DF/DR),见图1。引物序列见表2。
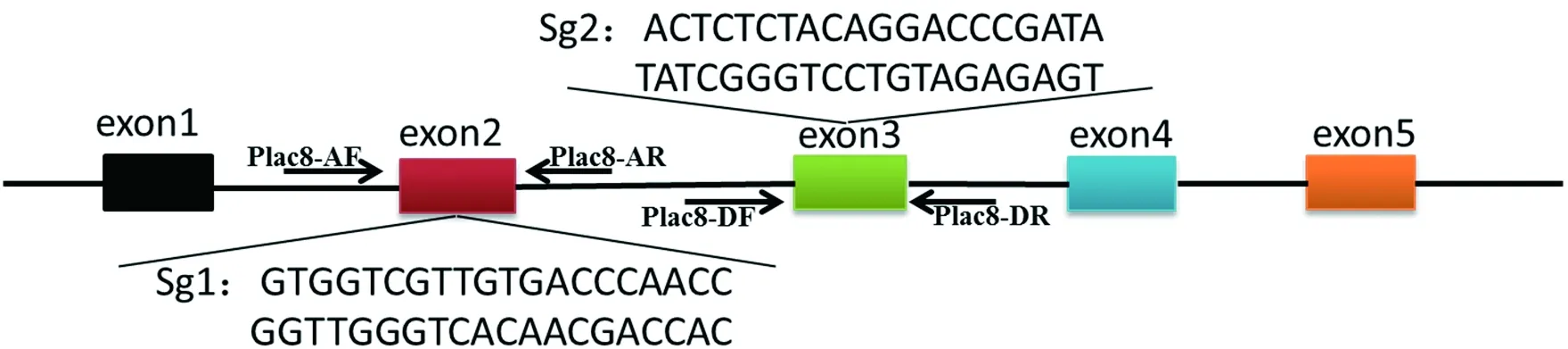
图1 PLAC8基因结构特征及sgRNA设计
1.3 PX458-sgRNA表达载体的构建和鉴定
选择PX458质粒作为载体,首先将含有质粒的保存菌液用LB液体培养基在37 ℃,180 r/min的摇床中培养14 ~ 16 h,用质粒小提试剂盒提取PX458质粒,测浓度,置于-20 ℃备用。
用Fast DigestBpiI酶切割质粒PX458,使其线性化,然后用2%的琼脂糖凝胶进行电泳,电泳后用凝胶成像系统检测片段大小及线性化效率,然后进行切胶回收,测浓度,置于-20 ℃备用。

表1 PLAC8 sgRNA引物序列

表2 PLAC8 敲除细胞系检测引物
将合成的sgRNA寡核苷酸单链梯度退火为双链,用T4 DNA连接酶与线性化的PX458连接,然后转化到stbl3感受态细胞中,挑取阳性单克隆菌落,将菌液做菌落PCR,将带有正确条带的菌落送测序验证是否构建成功。
1.4 细胞培养和细胞转染
293T细胞培养在含5% CO2,37 ℃恒温培养箱中。转染前24 h取对数期细胞以5×105/孔的细胞密度接种到6孔板中,细胞密度长到70%~80%时开始做转染。首先将细胞培养液换成不含血清的opti-MEM培养基饥饿30 min,然后将5 μL p3000试剂及2.5 μg的PX458-sgRNA重组质粒先后加入125 μL的opti-MEM中。与其同时,将7 μL的lipo3000试剂加入到另一管125 μL的opti-MEM中,边加边混合,静置5 min。然后将两管转染组分轻轻混合,再静置20 min后加入到事先饥饿好的细胞中。5 h后,换上含10%胎牛血清的完全培养液继续培养,24 h后观察荧光。
1.5 流式细胞仪分选
将准备分选的细胞用胰酶消化下来,1000 r/min离心5 min的条件下用PBS洗2次,然后取1 mL准备好的含1%胎牛血清的PBS将细胞吹打混匀,把细胞过滤膜分散成单细胞,收集在灭菌的15 mL离心管中,上机前用1 mL枪头轻轻混匀,准备好上机分选。上机后选择single模式,根据阴性对照调节电压,设置阳性门后,将带有绿色荧光的单个细胞打到事先加入200 μL含20%胎牛血清和2%双抗的完全培养基的96孔板中。
1.6 单细胞培养
将筛选出来的细胞养在含有20%血清和2%抗生素的完全培养基培养的96孔板中,约15 d便可以将细胞转到24孔板中,待细胞密度达到70%~80%就可以将细胞转入12孔板中,最后将细胞转到6孔板中继续培养。
1.7 测序检测
收取6孔板中的细胞提取基因组,送至武汉擎科生物公司进行测序。测序使用的引物为PLAC8-AF或PLAC8-DF。通过与野生型细胞基因组做比较,筛选出被编辑过的细胞,并将该细胞扩大培养后冻存,以便进行后续的实验研究。
1.8 Western Blot检测编辑效率及蛋白表达
提取被编辑细胞的蛋白质,以野生型293T细胞为对照,检测PLAC8及MAPK信号通路相关蛋白的表达。根据待检测蛋白分子量用SDS-PAGE凝胶配制试剂盒配制10%~15%的分离胶和5%的浓缩胶。浓缩胶30 mA、80 V跑50 min,分离胶30 mA、100 V跑1.5 h。电泳完毕后,200 mA、100 V湿法转膜1 h。 转膜后,用5%的脱脂奶粉将转好的膜封闭1 h,用TBST洗3次后,孵育一抗 (一抗稀释比例为1∶1000),4 ℃过夜。接着用TBST洗3次,每次10 min,室温下敷二抗1 h (二抗稀释比例为1∶10 000),TBST洗3次,每次10 min,TBS洗2次,每次5 min,用ECL显影,观察结果。所有的Western Blot实验均重复3次,并选取最具有代表性的结果进行呈现。
1.9 MTT检测细胞生长速度
取对数生长期被编辑的细胞系和野生型293T细胞,用PBS洗2次,用胰酶将细胞消化下来,1000 r/min离心5 min。加入1 mL 完全培养基将细胞吹散,置于含4 mL培养基的中皿里,混合均匀。取20 μL 细胞液置于1.5 mL 离心管中,再加入20 μL台盼蓝染液和60 μL PBS,用枪头混合均匀。取10 μL滴在血球计数板上,在显微镜下计数,重复3次,取平均数。
以2000个细胞/孔的密度将细胞铺在96孔板中,重复6个孔,铺5个96孔板。一天测一个96孔板。细胞贴壁后,在有细胞的孔中加入20 μL 5 mg/mL的MTT试剂,不用换液。4 h后除去孔内的完全培养基,加入150 μL DMSO。在酶标仪上震荡10 min,490 nm检测OD值,对所得数据进行统计分析。
2 结果与分析
2.1 敲除PLAC8基因的293T细胞株的构建
为了成功构建打靶载体,我们用T4 DNA连接酶将合成的sgRNA单核苷酸链退火连成双链,然后与线性化的PX458连接,转化到stbl3感受态中,37 ℃培养箱中过夜培养,挑取单克隆菌落,做菌落PCR后,将有正确条带的菌液进行测序。测序结果显示,sg-RNAS1、S2分别正确插入到PX458载体中。
接下来,我们用lipo3000转染试剂将构建好的两个打靶载体分别转染到293T细胞中,48 h后汞灯观察细胞内的绿色荧光,判断转染效率 (图2)。由图2可知,PX458-sgRNA-S1和PX458-sgRNA-S2这两个质粒都成功转染到293T细胞中,转染效率约为80%。

A: PX458-sgRNA-S1质粒转染细胞的荧光视野 (100×);b:PX458-sgRNA-S2质粒转染细胞的荧光视野(100×);c:PX458-sgRNA-S1质粒转染细胞的明场视野(100×);d:PX458-sgRNA-S2质粒转染细胞的明场视野 (100×)
图2敲除PLAC8基因的293T细胞株的构建
Figure 2 Construction ofPLAC8 knockout 293T cells a and c detection
of GFP via fluorescence microscopy after PX458-sgRNA-S1
transfection (green, PLAC8; 100×)
2.2 细胞株的鉴定
为了获得单一编辑的细胞,在载体成功转染至293T细胞后,利用流式细胞仪将转染后带有绿色荧光的细胞以每孔一个细胞的密度筛选至96孔板中培养,并通过基因组测序鉴定被编辑的细胞系。测序结果 (图3-a)显示,转染sgRNA2筛选出来两株细胞株基因组红色方框处发生了编辑,并出现了明显的套峰 (图3-b),分别命名为KD1和KD2。为了进一步验证该细胞是否成功编辑,使用错配酶T7E1进行验证,以KD2细胞株为例,结果显示其基因组能够被错配酶酶切成178 bp和299 bp两个小片段 (图 3-c)。以上实验共同证明KD1、KD2细胞系的基因组已被成功编辑。
为了进一步检测KD1、KD2细胞株基因组编辑的效率,对KD1、KD2细胞株的总蛋白进行了Western Blot实验,用野生型293T细胞做阴性对照。结果显示KD1、KD2细胞株中的PLAC8蛋白已经被完全敲除 (图 3-d)。
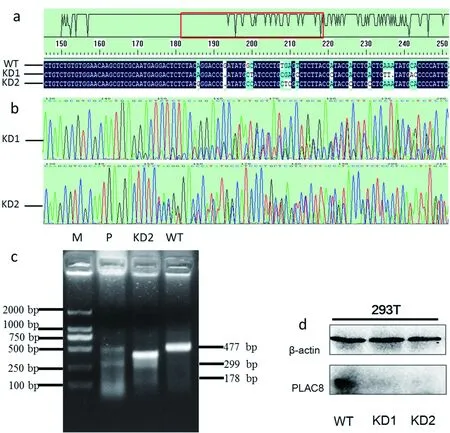
A: KD1、KD2与WT的基因组序列对比;b:KD1、KD2基因组测序峰图;c:以KD2为例,编辑细胞株错配酶验证结果,其中P为阳性对照 (Genloci CruiserTM基因敲除检测试剂盒中自带的阳性对照),KD2为被检测的敲除细胞系,WT为阴性对照,M为DNA Marker DL2000;d:Western Blot结果
图3 PLAC8敲除细胞株的验证
Figure 3 Identification of PLAC8 knockout cell line
2.3 PLAC8蛋白表达下调对293T细胞增殖的影响
将KD1、KD2细胞系与野生型293T细胞系,分别取对数期细胞以2000/孔的细胞密度接种到96孔板中,用5 mg/mL的MTT检测细胞的生长速度并分析数据。结果显示KD1、KD2细胞系的生长速度均明显慢于WT细胞系,具有统计学意义 (图 4-a)。

A:MTT结果图;b:与细胞增殖相关的4个基因在Plac8基因敲除细胞系 (KD2&KD1) 和野生型 (WT)中蛋白的表达量
图4 PLAC8表达下调对293T细胞增殖的影响
Figure 4 Down-regulated PLAC8 inhibits proliferation of 293T cells
为了进一步分析PLAC8 敲除影响细胞增殖可能的分子机制,通过Western Blot实验检测了一系列细胞增殖相关基因的蛋白表达水平的变化。结果显示 (图 4-b),能促进细胞生长和增殖的蛋白激酶B/AKT及核内原癌基因C-MYC表达下调,MAPK上游激酶RAF-1和胞外调节蛋白激酶ERK2表达上调。因此我们推测敲除PLAC8蛋白能改变ERK/MAPK信号通路中关键基因的表达变化,从而抑制细胞的增殖。
3 讨论
PLAC8是一种在多种癌细胞中表达并能调控肿瘤发生发展的蛋白。目前,国内外在细胞水平上研究PLAC8时都会用到基因编辑细胞系,其构建方式大多是使用CRISPR/Cas9基因编辑方法构建载体-sgRNA敲除重组质粒,再将该重组质粒导入病毒包装细胞中,收集病毒上清液转染细胞,然后用嘌呤霉素或者G418等抗生素进行筛选。通过提取基因组测序,筛选出敲除成功的细胞,再对敲除成功的细胞通过有限稀释法将细胞分成单克隆,最后在96孔板中培养。但是以上的方法操作起来十分繁琐,尤其是病毒实验需要成熟的技术和高要求的平台,有限稀释法需要大量的重复性工作,也不能确保能将细胞全部分为单克隆。
在本课题中,我们首先利用流式细胞术与CRISPR/Cas9基因编辑方法相结合来解决构建基因编辑细胞系过程中病毒包装转染效率低下及筛选目的细胞繁琐的瓶颈,以简便、高效、无毒的方法获得了PLAC8蛋白敲除人胚肾细胞系。后续的功能实验结果显示,在293T细胞系中,PLAC8蛋白表达下调会抑制细胞增殖,进一步的Western Blot实验结果显示,与野生型293T细胞相比,敲除PLAC8蛋白的293T细胞中,AKT,C-MYC基因的蛋白表达水平显著下降,ERK2,RAF-1基因的表达水平显著上升。AKT作为一种丝氨酸/苏氨酸蛋白激酶能够促进细胞增殖和生长,抑制细胞凋亡,促进细胞侵袭和转移[11-12]。RAF-1,ERK2和C-MYC是ERK/MAPK信号通路中的关键因子,RAF-1作为MAPK激酶激酶,激活后可以引起MAPK信号通路的活化,促使细胞增殖周期失调[13]。同时RAF-1的持续表达也可刺激抑制性因子P21cip1表达,使该因子与cdk2或cdk4结合增多而阻滞细胞周期,抑制细胞增殖[14]。ERK2是MAPK家族的一员,处于RAF-1的下游,调控信号通过ERK2从表面受体传导至细胞核,调控核内的转录因子C-MYC表达,继而影响基因转录,调控细胞增殖[14]。
综上所述,我们推测在293T细胞中PLAC8可能是通过刺激AKT以及RAF-1-ERK2-C-MYC信号级联通路调控细胞增殖。接下来我们还将通过后续蛋白水平上的实验进一步来验证其分子机制。本课题的研究一方面可将已证实可用的sgRNA用于后续编辑其他细胞系,另一方面也可以利用构建的敲除细胞系进行后续动物水平上的实验,如裸鼠荷瘤实验等,用以进一步研究PLAC8蛋白在肿瘤发生发展过程中的功能。
