基于共价有机骨架-MSPE-HPLC-UV法测定多环芳烃
2019-12-19胡碧清李倩莲严志明
胡碧清 - 叶 佳 李倩莲 - 庞 杰 严志明 -
(福建农林大学食品科学学院,福建 福州 350000)
多环芳烃(polycyclic aromatic hydrocarbons,PAHs)是重要的环境和食品污染物,通常来自有机物质的不完全燃烧,如煤、汽油、木材、肉类或其他有机食品[1]。其具有潜在的生物蓄积性,致突变、致癌和致畸性,对人类健康和环境构成潜在威胁[2-3]。广泛分布于环境和食品中的PAHs具有痕量水平的低浓度和低溶解性。因此,建立一种高效、灵敏的PAHs分析方法尤为重要。
目前,样品前处理技术主要包括固相萃取[4-5]、搅拌棒吸附萃取[6-7]、固相微萃取[8-9]、磁性固相萃取[10-12]等。磁性固相萃取是一种基于固相萃取的分散固相萃取技术,相比于其他前处理技术,只需使用少量的吸附剂和较短的平衡时间就能够实现低浓度的微量萃取,具有非常高的萃取能力和萃取效率。磁性固相萃取的核心是磁性纳米粒子,具有大的比表面积和独特的物理化学性质,特有的超顺磁性有助于通过磁铁方便地分离和收集。近年来,基于磁性纳米粒子的技术在分离领域中备受关注[13-15]。石墨烯、碳纳米管、金属—有机框架材料、分子印迹聚合物、共价有机骨架等材料用于制备磁性复合材料并应用于磁性固相萃取。
共价有机骨架材料(covalent organic framework,COFs)是一类完全由C、H、O、N等轻元素组成并且通过强共价键连接的有机晶体材料。COFs作为晶型多孔材料,具有低密度、高比表面积、良好的化学及热稳定性等特点而备受关注和研究[16-17]。在过去的10余年中,COFs在各个领域得到了广泛的应用,如气体存储与分离[18-20]、催化[21-23]、传感[24-25]、吸附剂[26-28]。近年来,COFs在样品前处理方面展现了巨大的应用潜力[29-30],尤其是磁性COFs材料可以克服因COFs材料密度轻及粒径小所引起的使用不便的问题。目前,磁性COFs的制备及在磁性固相萃取应用仍存在发展阶段,磁性COFs的制备存在操作繁琐、过程冗长、制备条件苛刻等问题。
试验分别制备磁性纳米颗粒Fe3O4及共价有机骨架材料COF-SCU1,再将酸化处理的Fe3O4与COF-SCU1通过静电吸附作用制备磁性共价有机骨架材料Fe3O4@COF-SCU1,并基于Fe3O4@COF-SCU1拟建立磁性固相萃取—高效液相色谱/紫外法分析大红袍茶汤中的8种PAHs,以期为检测环境和食品中的PAHs提供相关的理论依据。
1 材料与方法
1.1 材料与仪器
1.1.1 材料与试剂
1,3,5-苯三甲酰氯:纯度≥98%,上海阿拉丁生化科技股份有限公司;
甲醇、乙腈:色谱纯,国药集团上海化学试剂有限公司;
对苯二胺、三氯化铁、乙酸钠、乙酸乙酯、乙醇、乙二醇、丙酮、二氯甲烷:分析纯,国药集团上海化学试剂有限公司;
芴(Flu)、菲(Phe)、蒽(Ant)、荧蒽(Flt)、芘(Pyr)、苯并(a)蒽(BaA)、苯并(b)荧蒽(BbF)、苯并(a)芘(BaP)标准品:纯度≥99%,上海阿拉丁生化科技股份有限公司;
茶叶:大红袍,福建省武夷山市。
1.1.2 主要仪器设备
高效液相色谱仪:LC-20A型,配有紫外检测器,岛津-GL(上海)商贸有限公司;
电子天平:ME204/02型,梅特勒—托利多仪器(上海)有限公司;
超声波清洗器:KQ5200E型,昆明市超声仪器有限公司;
超纯水系统:Milli-Q IQ7000型,美国Merck公司;
氮吹仪:MG-2200型,上海虔钧科学仪器有限公司;
X射线粉末衍射(XRD):D8 Advance型,德国Bruker公司;
超导量子干涉装置磁力计(SQUID):PPMS-9型,美国Quantum Design公司;
物理吸附分析仪:ASAP 2020型,美国Micromeritics公司;
傅里叶变换红外(FT-IR)光谱仪:Nicolet 5700型,美国Nicolet公司;
透射电子显微镜(TEM):FEI Tecnai G20型,美国FEI公司;
扫描电子显微镜(SEM):FEI Inspect QUANTA 430型,美国FEI公司。
1.2 方法
1.2.1 Fe3O4@COF-SCU1的制备
(1) Fe3O4合成:根据文献[31]。将2.7 g三氯化铁和7.2 g乙酸钠溶解在100 mL乙二醇中,并将混合液置于反应釜中在200 ℃下反应8 h。反应结束后,用乙醇和超纯水清洗干净,于50 ℃真空干燥箱中干燥备用。
(2) COF-SCU1合成:根据文献[17]。将1.06 g 1,3,5-苯三甲酰氯溶解在30 mL乙酸乙酯中。将0.32 g对苯二胺溶解于15 mL乙酸乙酯,在0 ℃冰水浴条件下逐滴滴入1,3,5-苯三甲酰氯溶液中并在室温下搅拌24 h,反应结束后将所得的产物用乙醇和超纯水清洗干净,于50 ℃真空干燥箱中干燥备用。
(3) 制备Fe3O4@COF-SCU1:将2 g的Fe3O4溶解于0.1 mol/L的HCl溶液中,超声处理10 min,然后用超纯水清洗干净,并分散于20 mL超纯水中;将2 g的COF-SCU1分散于20 mL的超纯水中,超声处理10 min。将上述两种溶液混合,涡旋1 min,即可得Fe3O4@COF-SCU1。
1.2.2 色谱条件 色谱柱:Agilent C18(4.6 mm×150 mm,5 μm),柱温30 ℃,进样体积20 μL,流速1 min/mL。流动相:A为水,B为乙腈;梯度洗脱程序:0~5 min,60% B;5~11 min,60%~100% B;11~16 min,100% B;16~21 min,100%~60% B;21~25 min,60% B。PAHs的检测波长:Flu 210 nm,Phe 251 nm,Ant 251 nm,Flt 232 nm,Pyr 238 nm,BaA 287 nm,BbF 258 nm,BaP 295 nm。
1.2.3 混合标准溶液的配制 分别称取Flu、Phe、Ant、Flt、Pyr、BaA、BbF、BaP标准品,精确至0.000 1 g,用乙腈配制成1 000 μg/mL 的储备液。将8种标准储备液混合配制成200 μg/mL的混合标准溶液。用水逐级稀释质量浓度分别为4.00,2.00,1.00,0.50,0.25,0.10 μg/mL的标准溶液。将上述溶液置于4 ℃的冰箱中保存。
1.2.4 样品前处理 0.5 g大红袍茶叶加入120 mL开水冲泡3 min,茶汤用0.45 μm滤膜过滤,冷却至室温备用。
1.2.5 磁性固相萃取过程 萃取试验过程具体步骤如图1所示,将10 mg磁性吸附剂材料加入40 mL PAHs加标水样中,涡旋3 min使分析物与磁性材料充分接触。通过磁铁使吸附剂从水样中分离并去除上清液。加入2 mL二氯甲烷—乙腈(体积比1∶4)涡旋4 min,收集洗脱液并用温和的氮气吹至200 μL,用流动相定容至500 μL,经 0.22 μm微孔滤膜过滤,取20 μL于HPLC分析。
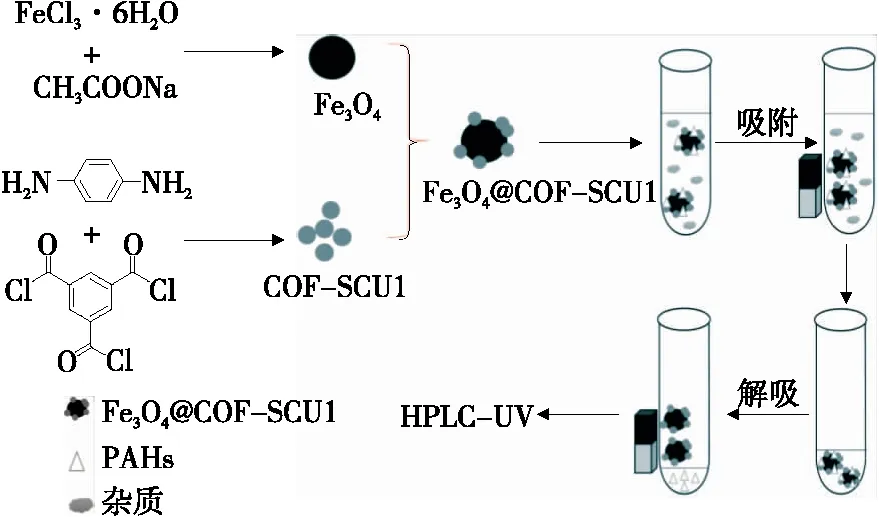
图1 Fe3O4@COF-SCU1的制备及MSPE-HPLC-UV法检测PAHs示意图
Figure 1 Schematic diagram of the preparation of Fe3O4@COF-SCU1 and the detection of PAHs by MSPE-HPLC-UV
2 结果与讨论
2.1 Fe3O4@COF-SCU1的表征
2.1.1 形貌表征 图2为SEM和TEM表征结果,揭示了材料的微观形态。如图2(a)所示,COF-SCU1为聚集状态,是由于大量的COF-SCU1团聚形成粒径约为200 nm 的小球形颗粒。从图2(b)可以观察到COF-SCU1黏附在Fe3O4周围和表面。从图2(d)可以观察到Fe3O4周围分布的COF-SCU1,进一步说明了COF-SCU1已经成功修饰在Fe3O4表面。
2.1.2 磁性能(VSM)分析 从磁滞曲线(图3)可知,Fe3O4和Fe3O4@COF-SCU1具有超顺磁性,Fe3O4饱和磁化值高达79.8 emu/g,Fe3O4@COF-SCU1饱和磁化值为55.4 emu/g,这种高饱和磁性可赋予其对磁铁的快速响应,利于收集和分离。
2.1.3 红外光谱(FTIR)分析 为了考察所制备材料的化学基团,分别对Fe3O4、COF-SCU1和Fe3O4@COF-SCU1进行了FTIR表征见图4。从COF-SCU1谱图可以观察到在3 378 cm-1处有很强的吸收峰,归属于—OH或—NH键的伸缩振动;在2 923 cm-1的峰为苯环C—H的伸缩振动峰;在1 713 cm-1的峰为苯环的特征吸收峰;在1 658 cm-1的峰为羧酸和酰胺C═O键的吸收峰;从Fe3O4谱图可以观察到578 cm-1处有强吸收峰,归属于Fe—O键伸缩振动引起的特征吸收峰,说明存在Fe3O4。从Fe3O4@COF-SCU1的FTIR图中清晰可见Fe3O4和COF-SCU1的特征峰,结果表明了Fe3O4@COF-SCU1为Fe3O4和COF-SCU1组成的复合材料。

图2 Fe3O4、COF-SCU1和Fe3O4@COF-SCU1的SEM图和TEM图
Figure 2 SEM images and TEM images of Fe3O4, COF-SCU1 and Fe3O4@COF-SCU1
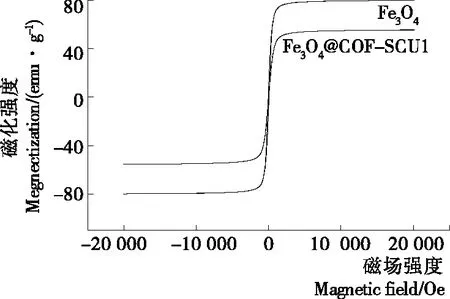
图3 Fe3O4和Fe3O4@COF-SCU1的磁滞曲线
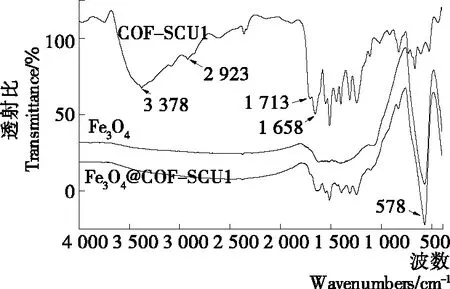
图4 Fe3O4、COF-SCU1和Fe3O4@COF-SCU1的FTIR图
2.1.4 X射线衍射(XRD)分析 采用XRD考察了Fe3O4、COF-SCU1和Fe3O4@COF-SCU1的晶体结构,结果见图5。Fe3O4分别在30.24°,35.48°,43.11°,57.00°,62.66°处有明显的特征衍射峰,与文献[31]相一致。COF-SCU1在26°处有较宽的衍射峰,说明其为无定型结构。从Fe3O4@COF-SCU1的XRD图可以观察到Fe3O4和COF-SCU1的特征峰均有出现,再次表明了Fe3O4@COF-SCU1为Fe3O4和COF-SCU1组成的复合材料。
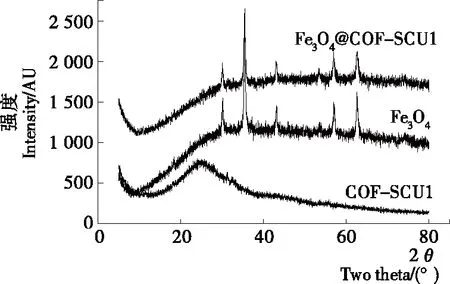
图5 Fe3O4、COF-SCU1和Fe3O4@COF-SCU 1的XRD图
2.1.5 比表面积(BET)氮气吸附脱附表征 通过氮气等温吸脱附,得Fe3O4的BET比表面积和孔体积分别为25.44 m2/g 和0.077 cm3/g。COF-SCU1的BET比表面积和孔体积分别为60.59 m2/g和0.47 cm3/g。
2.2 MSPE条件优化
2.2.1 Fe3O4与COF-SCU1配比的影响 Fe3O4@COF-SCU1主要通过疏水相互作用,π-π堆积作用力来萃取分析物,为获得最佳的萃取效果,分别制备了Fe3O4和COF-SCU1的质量比为5∶5,6∶4,7∶3,8∶2的吸附剂。如图6(a)所示,当质量比为5∶5时,观察到8种PAHs有较佳的回收率,且均在61%以上,其中BbF和BaP的回收率超过了90%。因此,后续试验使用的是基于Fe3O4和COF-SCU1的质量比为5∶5的吸附剂。
2.2.2 离子强度的影响 为了研究离子强度对萃取分析物的影响,使用NaCl来调节溶液的盐溶度。如图6(b)所示,当NaCl浓度从0.02 mol/L增加到0.06 mol/L时,分析物的回收率基本保持不变。而继续添加盐时,回收率均有所下降。这可能是添加的盐增加了溶液的黏度,影响了PAHs在吸附剂上的吸附动力学,因此后续试验中不添加NaCl。
2.2.3 吸附程序的优化
(1) 吸附剂用量对萃取效果的影响:考察了不同用量(5,10,15,20,25 mg)的吸附剂对萃取效率的影响。如图6(c) 所示,当吸附剂从5 mg增加到10 mg,PAHs回收率均有着显著的提高。其后随着吸附剂用量的增加,回收率基本保持恒定。因此,后续试验中吸附剂用量选择10 mg。
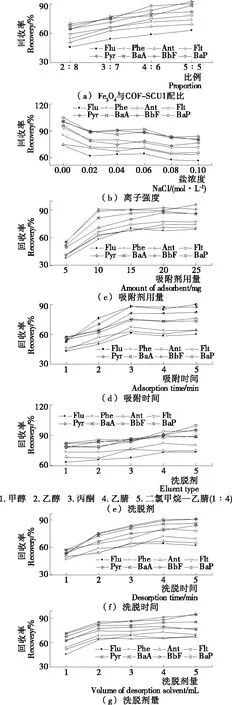
图6 磁性固相萃取条件优化
(2) 萃取时间对萃取效果的影响:如图6(d)所示,萃取3 min时8种PAHs即可达到吸附平衡。因此,在后续试验中萃取时间选择3 min。
2.2.4 洗脱程序的优化 选取甲醇、乙醇、丙酮、乙腈以及二氯甲烷—乙腈(体积比1∶4)5种洗脱剂进行了探索。从图6(e)可以观察到,二氯甲烷—乙腈(体积比1∶4)解析效果最好。因此,进一步优化了洗脱时间和二氯甲烷—乙腈(体积比1∶4)的用量,结果如图6(f)和(g)所示。最终选择2 mL的二氯甲烷—乙腈(体积比1∶4)作为洗脱溶剂,洗脱时间为4 min。
2.3 重复利用性
为考察材料的重复利用性,每次MSPE过程后,分别用1 mL的甲醇和水清洗吸附剂,再进行试验。经过5次试验,PAHs的回收率没有明显变化,说明Fe3O4@COF-SCU1具有良好的稳定性。
2.4 方法的线性范围与检出限
通过使用优化的MSPE条件确定方法的线性范围,灵敏度,验证方法的分析性能。配制一系列浓度的标准溶液,在最佳萃取条件下,经MSPE后进行HPLC-UV分析,如表1所示,8种PAHs均得到良好的线性关系,相关系数≥0.998 7。检测限(LODs,S/N=3)和定量限(LOQs,S/N=10)分别为0.10~0.40,0.33~1.34 ng/mL。
2.5 实际样品的分析
将建立的方法应用于大红袍茶汤中8种PAHs的分析检测。结果表明,大红袍茶汤中的Ant的含量为0.471 2 ng/mL,其余的PAHs未检出。分别添加浓度为2,10,20 ng/mL的PAHs混合标准溶液进行加标回收试验,重复3次。如表2所示,8种PAHs的平均回收率在74%~106%,相对标准偏差为1.20%~8.50%。图7为大红袍茶汤中分别加入浓度为2,10 ng/mL的PAHs混合标准溶液前后对比的色谱图。上述试验结果表明:该方法满足分析检测的要求,可用于检测低浓度的PAHs。
3 结论
通过简易的方法成功制备了Fe3O4@COF-SCU1并将其作为吸附剂,建立MSPE-HPLC-UV法检测大红袍茶汤中PAHs的分析方法。该方法操作简单,快速准确,充分利用了COF-SCU1的吸附能力以及MSPE的优势,避免了过滤等繁琐操作,且吸附剂可以快速方便地回收再利用,在同时富集萃取多种PAHs方面具有很好的应用前景。此外,研究出该技术的自动化萃取装置,实现在线磁性萃取进行实时检测分析,是该技术的进一步发展方向。
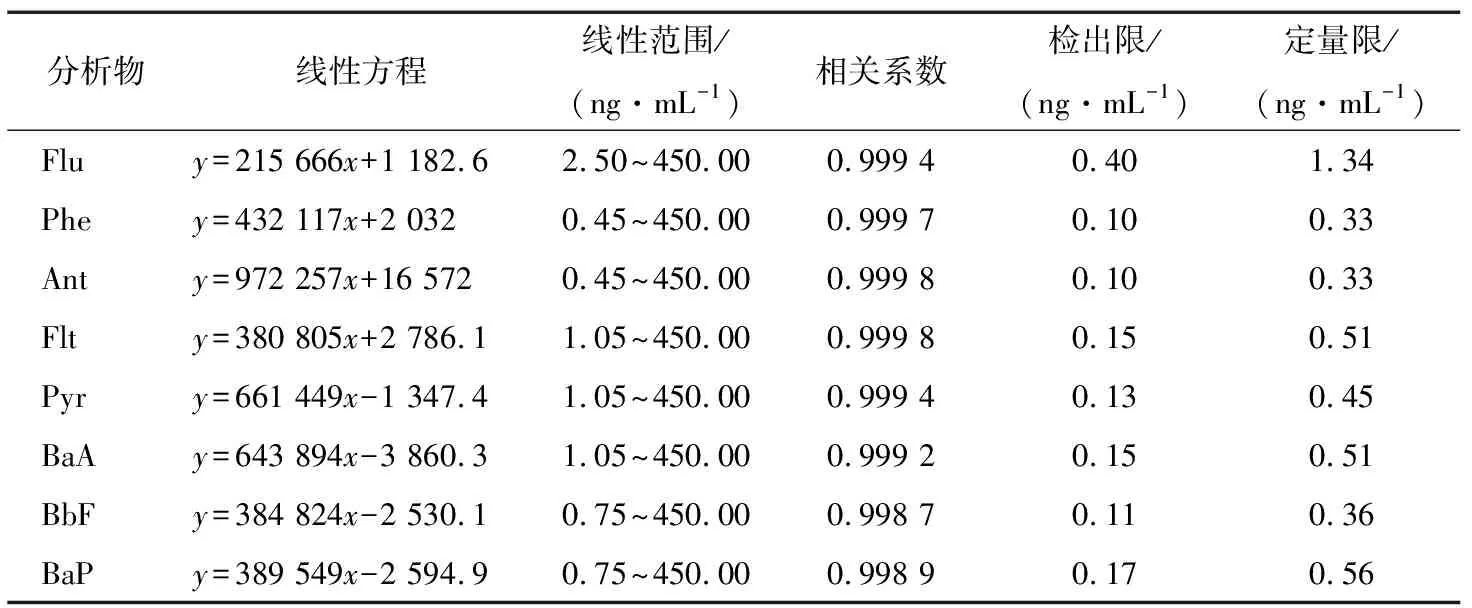
表1 方法的线性方程、线性范围、相关系数、检出限和定量限
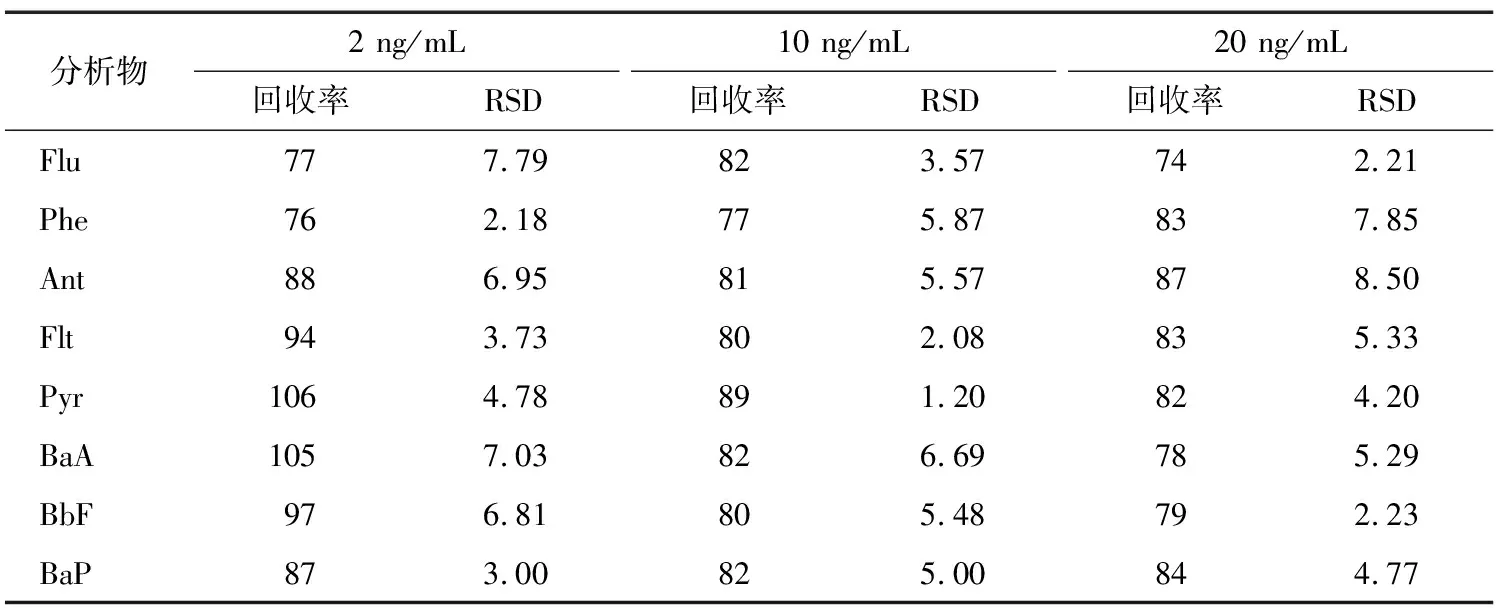
表2 大红袍样品中8种PAHs的加标回收率和相对标准偏差
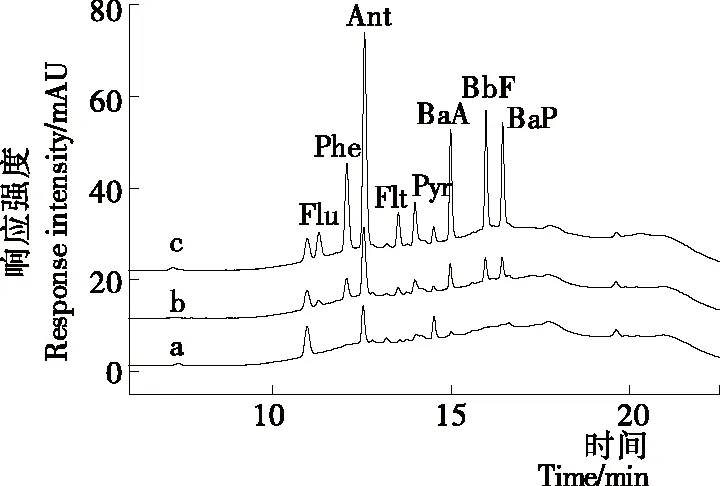
a. 没有加标 b. 加标2 ng/mL c. 加标10 ng/mL
