直系亲属中IgG4相关性疾病二例
2019-12-18戴彦苗徐宏伟许邹华陆喜荣钱陈洁王凯旋吴坚芳
戴彦苗 徐宏伟 许邹华 陆喜荣 钱陈洁 王凯旋 吴坚芳
昆山市中医医院消化内科,昆山 215300
【提要】 IgG4相关性疾病(IgG4-RD)是一组炎症性纤维化疾病,主要表现为一个或多个器官受累肿胀、纤维化,多伴有血清IgG4水平升高。由于几乎可以累及全身各个器官,容易误诊。此病存在一定的遗传倾向,但确切的发病机制不明,可能与人类白细胞抗原等有关。目前国内外关于此病多为散发病例报道,家族性报道少见,本文报道近期诊断的直系亲属中罹患IgG4-RD患者2例,两者血IgG4水平升高,经糖皮质激素治疗后症状改善,目前随访2年余病情稳定。
病例1 患者男性,63岁。因“中上腹胀满不适伴乏力、纳差半月”入院。4年前患者因活动后胸闷、气促在上海某医院行胸部CT检查发现纵隔及肺门4、5、7、8、10 R/L组淋巴结增大,PET-CT未见肺部异常放射性浓聚,胸腔镜淋巴结活检未见恶性细胞,未予特殊处理。平日活动后略有气促、喉中哮鸣音。否认家族遗传病史。体检:皮肤、巩膜无黄染,心肺及腹部未见异常。实验室检查:肝功能ALT 74.5 U/L,AST 48.8 U/L,ALP 132.1 U/L,GGT 569.0 U/L,ALB 32.8 g/L,GLOB 61.3 g/L;肿瘤标志物AFP、CEA、CA19-9均正常;凝血功能、血常规无明显异常。B超见胆总管及肝内胆管扩张,胰头处实质性占位。胸部+全腹部增强CT见纵隔淋巴结肿大;胰头部点状钙化灶,胰腺呈腊肠样,门脉期延迟强化,胰管稍扩张,肝内胆管及胆总管扩张;后腹膜纤维化。上腹部增强MR+MRCP见胰管扩张,肝内胆管及胆总管扩张,胆总管下段狭窄。后查免疫球蛋白IgG 30.9 g/L(正常值7~16 g/L),IgM 0.3 g/L(正常值0.4~2.3 g/L),IgG4 46.3 g/L(正常值0.03~2 g/L);ANA、ENA无明显异常。行EUS见胰头部一2.4 cm×2.4 cm混合回声肿块,边界不清,胆总管扩张,细针穿刺(FNA)标本病理检查未见异型细胞。既往纵隔淋巴结切片行免疫组织化学染色结果IgG4(+),>30个/高倍视野(图1)。综合影像、血IgG4水平及相关病理结果,诊断为IgG4相关性疾病(IgG related disease, IgG4-RD),累及胰腺、胆管、后腹膜、纵隔淋巴结。予泼尼松40 mg,1次/d,口服,并辅以补钙、护胃治疗。20 d后复查肝功能ALT 70.8 U/L,AST 27.1 U/L,ALP 96.9 U/L,GGT 378.5 U/L,ALB 34.8 g/L,GLOB 37.9 g/L。患者症状好转后出院。门诊激素逐渐减量,3月后复查IgG4 7.75 g/L,上腹部增强MR+MRCP见胰管、胆管无扩张,胆管狭窄,胰腺肿胀明显减轻,腹膜后纤维化好转(图2)。目前治疗随访中,血清IgG4逐步升高,但影像及其他生物化学均无异常。
病例2 患者男性(与病例1是亲兄弟),72岁。因“身、目、尿黄半月”入院。入院前半月出现身、目、尿黄,伴有腹泻、里急后重感,近1年来体重下降5 kg左右。否认家族遗传病史。体检:皮肤、巩膜黄染,腹软、无压痛、反跳痛,腹部未触及包块。肝脏肋下未触及,脾脏未触及。实验室检查:ALT 18.2 U/L,AST 117.4 U/L,ALP 379.5 U/L,GGT 419.9 U/L,ALB 27.5 g/L,GLOB 42.5 g/L,TB 302.6 μmol/L,DB 168.7 μmol/L;IgG4 29 g/L;AFP、CEA、CA19-9均正常。全腹部增强CT见胆总管及肝内胆管扩张,胰体尾部胰管扩张,考虑胰腺癌可能。MRCP见肝内胆管、胆总管及胰管扩张,胆总管下段狭窄。EUS见胰头部肿块,胆总管扩张,胆管壁增厚(图3),FNA穿刺标本未见恶性细胞。结合CT、MR、EUS,诊断为IgG4-RD,累及胰腺、胆管。予强的松30 mg,1次/d,口服,后逐渐减量,10 mg维持。4月后复查MR,胆管及胰管无扩张,胰腺肿胀消退,胰头部未见肿块(图4)。复查IgG4为8.14 g/L。目前治疗随访中,血清IgG4逐步升高,但影像及其他生物化学指标均无异常。
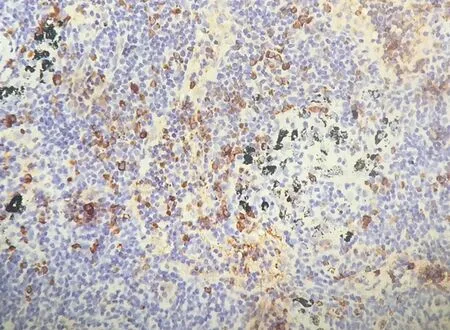
图1 淋巴结免疫组织化学染色可见大量IgG4淋巴细胞浸润
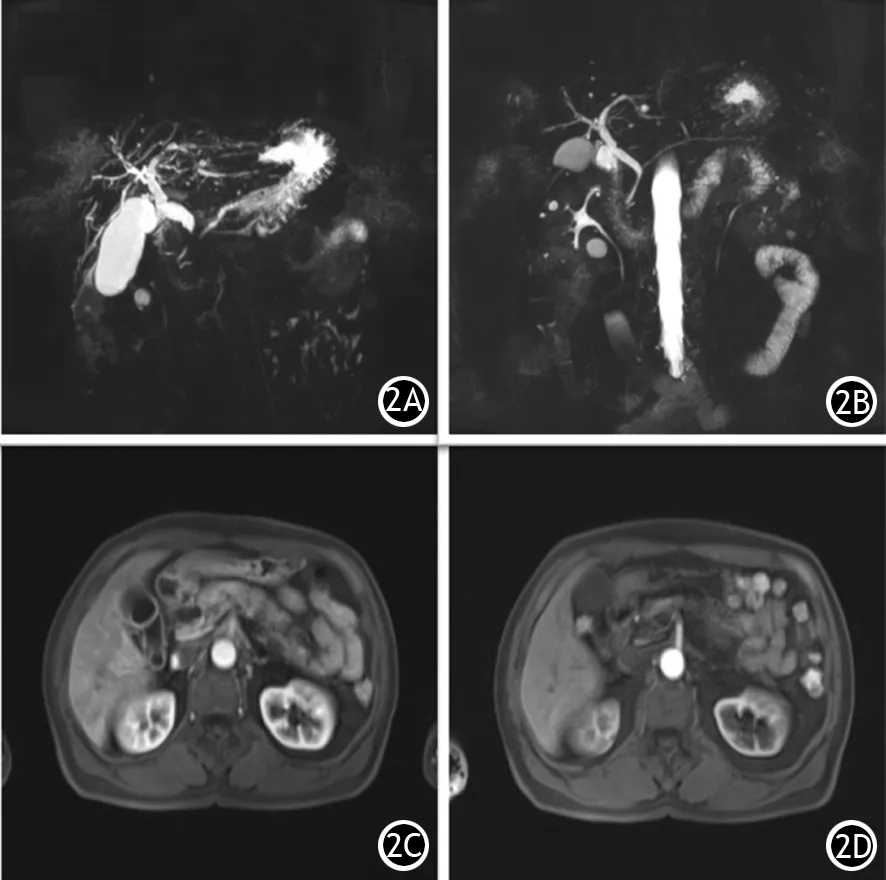
图2 激素治疗前MRCP示胆胰管扩张,下段狭窄(2A);治疗后胆胰管不扩张(2B);治疗前MR示腹膜后纤维化(2C);治疗后腹膜后纤维消失(2D)
讨论IgG4-RD的认识最早起源于自身免疫性胰腺炎(autoimmune pancreatitis,AIP)。1995年日本学者Yoshida等[1]首次提出了AIP的概念,并指出该病的发病机制和自身免疫因素相关,对糖皮质激素治疗效果较好,但较易复发。2001年Hamano等[2]发现血清IgG4的水平升高与AIP存在相关性。2003年Kamisawa等[3]证实了在胰腺及其他受累组织中存在大量IgG4阳性的浆细胞和纤维化,故而认为AIP不单是胰腺的病变,而是一种系统性疾病在胰腺的表现,进而提出了IgG4相关性自身免疫性疾病的概念。到2011年此病才被统一命名为IgG4-RD。
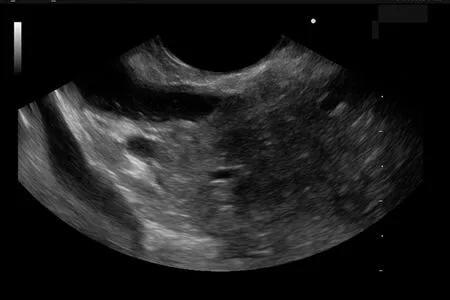
图3 EUS见胆总管扩张,胆管壁增厚,胆总管下端低回声占位

图4 治疗前MR示胰头部肿胀,疑似占位(4A);治疗后胰头部肿块缩小(4B);治疗前MRCP示胆总管中下段节段狭窄,上段扩张,胰管扩张(4C);治疗后胆胰管无扩张,胆管狭窄消失(4D)
AIP是IgG4-RD中最常见的一种,也是最早被认识的疾病,其人群发病率0.8/10万,约占慢性胰腺炎的5%左右[4]。多数患者可出现多个器官的受累,Sah和Chari[5]报道多器官受累的患者占60%~90%。目前此病已逐渐被临床医师所认识,我国也出台了AIP的相关诊疗共识,指导临床诊断与治疗[6]。IgG4-RD作为一个系统性疾病,可累及全身多个组织器官,包括胰腺、泪腺、涎腺、垂体、甲状腺、肺、主动脉、冠状动脉、肝胆、肾脏、前列腺、皮肤及淋巴结等。其典型的病理表现为富含IgG4阳性的淋巴细胞和浆细胞浸润、席纹样纤维化、闭塞性静脉炎等。临床上表现为受累器官肿胀、增生,压迫周围脏器及功能障碍等,多伴有免疫球蛋白升高,尤其是IgG4水平升高。由于其临床表现多样,容易被漏诊和误诊。国际上目前诊断多参照 2012年日本研究委员会提出的综合分类标准[7]:(1)一或多个器官出现弥漫性或局限性肿胀,或肿块;(2)血清IgG4≥1 350 mg/L;(3)组织病理学检查:①显著的淋巴细胞、浆细胞浸润和纤维化;②IgG4阳性浆细胞浸润,IgG4或IgG阳性细胞占比>40%,且IgG4阳性浆细胞>10个/高倍视野。确定诊断:(1)+(2)+(3);可能诊断:(1)+(3)或(1)+(2)。所有患者均需排除结核、肿瘤、血液系统疾病及其他自身免疫性疾病。
IgG4-RD好发于60岁左右中老年男性,但目前确切的病因和发病机制仍不清楚。研究认为此病有一定的遗传倾向,人类白细胞抗原(HLA)-DRB1*04:05和DQB1*04:01可能和AIP的发病有关,不同的HLA-DR和HLA-DQ的基因亚型与不同脏器受累有关。同时Fc受体样基因3(FCRL3)、细胞毒性T淋巴细胞相关抗原4(CTLA-4)、Toll样受体4(TLR4)和蛋白酪氨酸磷酸酶非受体型22(PTPN22)也被认为与IgG4-RD有关[8],但目前关于此病的家族性报道少见。2013年Watanabe等[9]报道了两个亲兄弟罹患AIP,并对其HLA-DR和HLA-DQ的基因亚型进行了研究分析,并没有发现存在与之前报道的高表达的HLA类型。本研究的2例患者为亲兄弟,有相同的遗传背景,但临床表现各不相同,而该家族中其他4位兄弟姐妹均未有发病,反映了IgG4-RD可能是受多种因素影响的一类疾病。2例患者的血清IgG4水平在随访期间都有不同程度的升高,但临床未见复发表现,这是否是这种家族性发病的特征尚有待进一步随访观察。
利益冲突所有作者均声明不存在利益冲突
