利用BAC/galK系统构建缺失潜伏相关转录物启动子LAP1的伪狂犬病突变病毒
2019-12-16张丽荣童光志
陈 静,张丽荣,童 武,叶 超,童光志,郑 浩
(中国农业科学院上海兽医研究所,上海 200241)
伪狂犬病(pseudorabies,PR)是一种由伪狂犬病病毒(Pseudorabies virus,PRV)引起的急性、热性传染病。PRV能感染多种家畜和野生动物,如猪、牛、羊、犬、鼠等。猪是PRV的唯一自然宿主,对其他动物均引起致死性感染。感染能引起仔猪高死亡率,造成母猪流产、产死胎和木乃伊胎,公猪多表现为睾丸肿胀、萎缩、种用性能降低等危害[1,2],给养猪业可造成极大的经济损失。通过接种基因缺失弱毒疫苗和相应的鉴别诊断方法,我国一些猪场已控制或根除了伪狂犬病,但是大部分地区仍存在该病的流行,特别是2011年末以来PRV变异株在我国的爆发流行,严重影响着养猪业的健康发展[3-6]。
PRV在感染耐过猪中能形成潜伏感染。潜伏感染期间病毒基因组不复制,也不表达病毒蛋白,仅大量转录病毒潜伏相关转录物(latency-associated transcripts,LATs)[7]。LATs由大的潜伏相关转录本(large latency transcripts,LLT)分子剪接加工形成,尚未发现LATs存在编码产物[8]。病毒基因组虽然在潜伏期间不复制,但是仍然保留其全部功能。潜伏感染再激活后,基因组重新复制并释放病毒粒子感染其他动物[9,10],这也是猪伪狂犬病难以根除的主要原因。
细菌人工染色体(bacterial artificial chromosome,BAC)是可以插入100~300 kb DNA片段的克隆载体。PRV感染性BAC克隆,是将PRV全基因组克隆至BAC载体,以质粒的形式在大肠杆菌中稳定的遗传增殖[11]。提取BAC克隆载体转染细胞,能获得感染性病毒。由于克隆中病毒序列较长,突变BAC克隆中的病毒序列,需要借助一些特定的筛选方法。galK(半乳糖激酶)正负筛选系统是操作BAC克隆的常用方法。galK与galK-宿主菌结合,置换下需要突变病毒序列。在含有半乳糖作为碳源的平板上选择galK+的重组细菌,然后负筛选,以含有特定DNA突变的片段取代galK基因,在甘油作为碳源,含有2-脱氧-D-半乳糖(2-Deoxy-D-galactose,DOG)的平板上筛选。DOG能被galK磷酸化为有毒的不可代谢中间体2-脱氧-半乳糖-1-磷酸,从而抑制galK+细菌生长,在筛选平板上长出的菌落即为galk基因被目的片段置换的克隆菌[12]。
本实验室已经成功构建PRV JS-2012株的BAC克隆,即pBAC-JS2012。将pBAC-JS2012电转入大肠杆菌SW102菌株中获得SW102-BAC,再结合galK正负筛选系统,成功构建了PRV感染性细菌人工染色克隆体操作平台[13],可以操作PRV的任何基因。本研究利用已构建的操作平台,获得缺失LLT启动子LAP1并添加cHS4的BAC载体pBAC-JS-cHS4-△LAP1b。通过与质粒pcDNA3.1-cre共转染Vero细胞后,获得不含BAC载体的突变病毒vJS-cHS4-△LAP1b,分析突变病毒的体外生物学特性。通过RT-PCR探究突变病毒感染Vero细胞后LLT的表达特征,进而研究该突变病毒对病毒复制、致病性及LLT调控的非编码RNA转录的影响。
1 材料和方法
1.1 病毒、细胞、载体和菌株PRV JS-2012由本实验室分离鉴定并保存[14];Vero细胞、pGalK质粒、pBluescriptⅡSK(+)载体、含JS-2012株BAC克隆pBAC-JS2012的大肠杆菌菌株SW102-BAC均由本实验室保存[13]。
1.2 工具酶和主要试剂限制性内切酶和T4 DNA连接酶购自NEB公司;Lipofectamine 3000 Transfection Kit购自Invitrogen公司;RevertAid First Strand cDNA Synthesis Kit购自ThermoFisher公司;胎牛血清(fetal bovine serum,FBS)、OPTI-MEM、0.25%Trypsin-EDTA(1×)和Penicillin Streptomycin购自Gibco公司;DMEM购自Sigma公司;胶回收试剂盒购自广州东盛科技有限公司;质粒小提试剂盒购自Omega公司;质粒中提试剂盒购自Macherey-Nagal公司;PCR试剂购自TaKaRa公司;M63 Broth、D-Biotin、Chloramphenicol、D-Galactose和2-Deoxy-D-galactose购自生工生物(上海)有限工程公司;大肠杆菌DH5α感受态细胞、氨苄青霉素购自北京天根公司。
1.3 序列合成和引物设计含绝缘子cHS4序列的pUC57载体,由金唯智生物科技公司合成。根据已发表的伪狂犬病病毒JS-2012基因组序列(GeneBank登录号:KP257591.1),设计缺失启动子LAP1的左右同源臂L1和R2引物:LATL1F/LATL1R、LATR2F/LATR2R。结合载体pGalK序列,设计扩增含同源臂的galK引物:LatgalKF/LatgalKR;设计正筛选检测galK是否成功插入SW102-BAC中的引物:LatDF/LgalKdown;设计检测负筛选是否成功的引物:PL1F/PR2R。根据LLT的ORF1和ORF2序列设计RT-PCR的引物:LLTORF1-RT和LLT-ORF2-RT,检测ORF1 cDNA的引物:651F/946R,以及检测ORF2 cDNA的引物:393F/697R。以上引物均由上海桑尼生物科技有限公司合成(表1)。

表1 本研究设计引物信息Table1 Primers used in this study
1.4 转移载体pBluescript-L1-cHS4-R2的构建以JS-2012为模板,使用LATL1F/LATL1R和LATR2F/LATR2R引物分别扩增同源臂左臂L1和右臂R2。用XhoⅠ和BamHⅠ酶切左臂L1,用BglⅡ和XbaⅠ酶切右臂R2,用XhoⅠ和XbaⅠ酶切pBluescriptⅡSK(+)载体,用XhoⅠ和BamHⅠ酶切载体pUC57-cHS4。T4切胶回收的pUC57-cHS4大片段和左臂L1用T4 DNA连接酶进行连接后,转化DH5α感受态细胞,提取载体pUC57-L1-cHS4。用XhoⅠ和BglⅡ酶切pUC57-L1-cHS4载体,回收目的片段L1-cHS4。将酶切后的pBluescriptⅡSK(+)载体、右臂R2、L1-cHS4通过T4-DNA连接酶连接,构建重组质粒pBluescript-L1-cHS4-R2,使用XhoⅠ和XbaⅠ进行初步酶切鉴定。
1.5 galK正负筛选系统
1.5.1 扩增galK基因 以载体pGalK为模板,使用引物LatgalkF/LatgalkR扩增galK基因片段并胶回收扩增产物。
1.5.2 galK正筛选 首先复苏含有BAC载体的JS-2012菌液SW102-BAC,制作正筛选电转感受态细胞,将galK片段电转化至感受态细胞中,电击条件:1800 V、200 Ω、20 μF、1 mm。用M9盐溶液洗3遍后涂正筛选平板,32℃恒温培养箱中培养3 d;挑取单菌落于氯霉素培养基中,32℃、200 r/min培养16 h;用引物LatDF/Lgalkdown检测菌液,选择阳性菌液并涂麦康凯平板进行筛选、纯化,挑取麦康凯平板上的红色单菌落于氯霉素平板上继续纯化;对纯化后的菌液SW102-BAC-galK进行鉴定和测序,测序成功的菌液保存于-80℃。
1.5.3 galK负筛选 以构建的负筛选载体pBluescript-L1-cHS4-R2为模板,使用引物LATL1F/LATR2R扩增目的片段L1-cHS4-R2。复苏正筛选获得的含galK的阳性菌液SW102-BAC-galK,制作负筛选感受态细胞。将目的片段L1-cHS4-R2电转化至感受态细胞中,电击条件:1800 V、200 Ω、20 μF、1 mm;用M9盐溶液洗3遍后涂负筛选平板,32℃恒温培养箱中培养3 d;挑取单菌落于氯霉素培养基中,32℃、200 r/min培养16 h;用引物PL1F/PR2R检测菌液,选取阳性菌液涂氯霉素平板进行纯化;对纯化后的菌液SW102-BAC-cHS4-△LAP1b进行鉴定和测序,测序成功的菌液保存于-80℃。提取菌液SW102-BAC-cHS4-△LAP1b的BAC DNA:pBACJS-cHS4-△LAP1b,-20℃保存备用。
1.6 病毒拯救
1.6.1 拯救病毒vJS-cHS4-△LAP1b-BAC 用Lipofectamine 3000 Transfection Kit将2 μg pBACJS-cHS4-△LAP1b转染至Vero细胞中,待细胞80%病变时收取上清,获得病毒vJS-cHS4-△LAP1b-BAC,保存于-80℃。
1.6.2 拯救病毒vJS-cHS4-△LAP1b 用Lipofectamine 3000 Transfection Kit将2 μg pBAC-JS-cHS4-△LAP1b和1 μg pcDNA3.1-cre共转染至Vero细胞中,待细胞80%病变时收取上清,通过三轮空斑纯化,获得不含绿色荧光、删除BAC载体的突变病毒vJS-cHS4-△LAP1b,保存于-80℃。
1.7 病毒体外生物学特性分析
1.7.1 病毒一步生长曲线 将亲本病毒JS-2012和突变病毒vJS-cHS4-△LAP1b按照MOI=1的接毒量稀释,分别接种于长满Vero细胞的T25细胞瓶中,37℃孵育1 h后,弃掉病毒液,用0% DMEM洗2遍,加入5 mL 2%FBS的DMEM置于37℃、5%CO2培养箱中培养。在接毒后第4、8、12、16、20、24、28、32 h收取上清病毒液500 μL,并补加500 μL 2% FBS的DMEM。将不同时间点的病毒液进行10倍梯度稀释,从10-1稀释至10-8,并接种于长满Vero细胞的96孔板中,每个稀释度接8个孔,并做两个重复。接毒后的96孔板放置于37℃、5%CO2培养箱中。4 d后记录病变孔数,用Reed-Muench法计算每个病毒的TCID50,并通过GraphPad软件绘制该病毒的一步生长曲线图。
1.7.2 病毒空斑实验 首先配制空斑实验所需要的2%低熔点凝胶培养基:高压灭菌后的低熔点凝胶与2×MEM按照1∶1比例混匀,并加入2%FBS,混匀后置于37℃培养箱备用。亲本病毒JS-2012和突变病毒vJS-cHS4-△LAP1b按照 MOI=1的接毒量稀释后,分别接种于长满Vero细胞的六孔板内,于37℃培养箱孵育1 h;弃去病毒液,用PBS洗两遍,每孔加入2.5 mL配置好的2%低熔点琼脂培养基,室温凝固;倒置于37℃、5%CO2培养箱中,70 h后用龙胆紫染色,观察空斑大小和形态。
1.8 vJS-cHS4-△LAP1b LLT表达特征的RT-PCR检测按照MOI=0.5接毒量,将亲本病毒JS-2012和突变病毒vJS-cHS4-△LAP1b分别接入长满Vero细胞的六孔板中,待细胞病变打到80%,提取细胞的总RNA进行DNaseⅠ处理。用反转录引物LLT-ORF1-RT和LLT-ORF2-RT通过RT-PCR将RNA反转录为cDNA。cDNA用651F/946R引物检测LLT ORF1,用393F/697R引物检测LLT ORF2。
2 结果
2.1 pBluescript-L1-cHS4-R2载体的构建及鉴定以JS-2012为模板,PCR获得204 bp的左臂L1,以及269 bp的右臂R2。将酶切后的左臂L1和载体pUC57-cHS4连接构建载体pUC57-L1-cHS4。酶切载体pUC57-L1-cHS4和pBluescriptⅡSK(+),胶回收酶切产物,将酶切后的pBluescriptⅡSK(+)、R2和L1-cHS4连接成为载体pBluescript-L1-cHS4-R2。将酶切鉴定成功的质粒送公司测序,测序结果正确的载体-20℃保存。
2.2 正筛选鉴定结果以pGalK序列为模板,扩增galK基因片段,大小为1336 bp。将galK片段电转入SW102-BAC中。从正筛选平板上挑取单菌落20个,用引物LatDF/Lgalkdown进行菌液PCR鉴定。电泳结果显示,有9个与预期大小一致的阳性克隆菌液SW102-BAC-galK(图1)。将阳性克隆菌液涂麦康凯平板进行筛选、纯化,挑取麦康凯平板上的红色单菌落涂氯霉素平板纯化后送公司测序,最终测序结果与预期序列相符合。
2.3 负筛选鉴定结果L1-cHS4-R2片段电转入SW102-BAC-galK菌后,将转化菌液涂负筛选平板。从负筛选平板上挑取单菌落,用引物PL1F/PR2R进行菌液PCR鉴定。电泳结果显示,有13个与预期大小一致的阳性克隆SW102-BAC-cHS4-△LAP1b(图2)。将阳性克隆的菌液涂氯霉素平板纯化后送公司测序,最终测序结果与预期序列相符合。
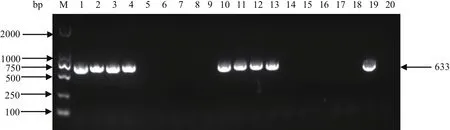
图1 galK正筛选鉴定结果Fig.1 PCR identification of positive selection

图2 负筛选鉴定结果Fig.2 Identification of negative selection
2.4 突变病毒的筛选及纯化将突变病毒pBAC-JS-cHS4-△LAP1b转染至Vero细胞中,获得病毒vJS-cHS4-△LAP1b-BAC,该突变病毒感染Vero细胞后表达绿色荧光蛋白。将突变病毒pBAC-JS-cHS4-△LAP1b和pcDNA3.1-cre共转染至Vero细胞后,经过三轮空斑纯化,最终获得不含绿色荧光并完全删除BAC载体的突变病毒vJS-cHS4-△LAP1b(图3)。
2.5 病毒一步生长曲线亲本病毒JS-2012和突变病毒vJS-cHS4-△LAP1b接种Vero细胞后,根据不同时间点的病毒滴度绘制病毒一步生长曲线(图4)。结果显示,突变病毒vJS-cHS4-△LAP1b的复制速度与亲本病毒JS-2012基本一致。
2.6 病毒空斑大小及形态亲本病毒JS-2012和突变病毒vJS-cHS4-△LAP1b接种Vero细胞后,用2%低熔点琼脂培养基培养70 h后进行龙胆紫染色,观察亲本病毒JS-2012和突变病毒vJS-cHS4-△LAP1b空斑大小和形态(图5)。结果显示,突变病毒vJS-cHS4-△LAP1b与亲本病毒JS-2012相比,空斑大小和形态基本一致。
2.7 病毒LLT RT-PCR检测结果亲本病毒JS-2012和突变病毒vJS-cHS4-△LAP1b分别感染Vero细胞后,提取细胞总RNA,将其反转录为cDNA。对LLT的ORF1和ORF2 PCR检测结果表明,亲本病毒和突变病毒感染Vero细胞后LLT ORF1(大小为296 bp)和ORF2(大小为305 bp)均能表达(图6)。

图3 突变病毒筛选纯化图Fig.3 Screening and purification of mutant virus

图4 亲本病毒与突变病毒一步生长曲线Fig.4 Single-step growth curves of the parental virus and mutant virus
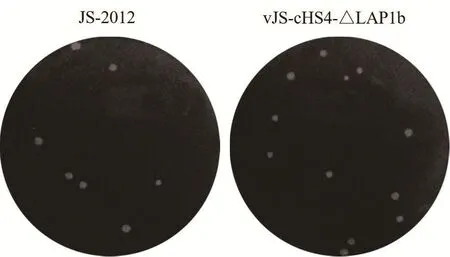
图5 亲本病毒和突变病毒空斑大小和形态Fig.5 Plaque size and morphology of the parental virus and mutant virus

图6 亲本病毒和突变病毒LLT的表达特征Fig.6 Expression feature of LLT of the parental virus andmutant virus
3 讨论
PRV具有的潜伏感染及再激活的特性是其能够在猪群中持续存在的重要原因之一[9],但是具体调控机制尚不清楚。由于PRV基因组庞大,在构建突变病毒时不易操作,很大程度上阻碍了PRV相关病原生物学特性和调控机制研究的进展[15]。
本研究基于实验室前期已经成功构建的pBACJS2012载体和galK正负筛选系统技术[13],操作简单、快速高效、准确率高,不引入其他外源序列。通过将JS-2012基因组插入BAC载体,实现了在原核生物系统对PRV基因组进行拷贝的扩增、提取和保存。同时进一步利用galK系统,实现了在原核生物系统中对PRV基因组特定基因组件进行替换、删除和插入。本研究将PRV LLT的启动子LAP1成功缺失,并且利用真核细胞系统的cre酶,获得不含BAC载体的突变病毒vJS-cHS4-△LAP1b,继而可以研究该突变病毒感染Vero后,PRV LLT的表达特征。对该突变病毒添加绝缘子cHS4是为了抑制启动子上游某些调控序列对下游启动子活性的影响。
通过对突变病毒的体外生物学特性分析,我们发现突变病毒与亲本病毒JS-2012空斑形态和大小基本一致。一步生长曲线也表明突变病毒的复制速度与亲本病毒JS-2012基本一致。空斑实验和一步生长曲线均表明,突变病毒与亲本病毒JS-2012体外生物学特性基本一致。由此可知,将PRV病毒LLT启动子LAP1缺失后,对于病毒在体外的复制和扩散能力没有显著影响。不过突变病毒在动物体内的感染致病特性是否发生改变,还需要相应的体内实验来进行验证。
为探究LAP1对LLT相关转录本表达情况的影响,本研究将突变病毒和亲本病毒分别感染Vero细胞后,对潜伏相关转录本LLT的ORF1和ORF2表达情况进行了RT-PCR检测。结果表明,同亲本病毒一样,突变病毒依然能检测到LLT的ORF1和ORF2。可见,在体外裂解性感染中,缺失LLT启动子LAP1并不能关闭LLT的表达。但LAP1的缺失是否影响LLT的表达量?后续我们将进行荧光定量分析,确定该突变病毒vJS-cHS4-△LAP1b与亲本病毒JS-2012在LLT表达量上的差异,以及LLT上非编码miRNA的表达情况,从而阐明启动子LAP1对LLT转录本表达特征的影响机制,以及对病毒潜伏感染能力的影响。
