石墨相氮化碳/二氧化锡复合材料的制备及光催化性能
2019-12-11武月桃王续峰刘艳丽
宁 湘 武月桃 王续峰 刘艳丽*,
(1湖南大学材料科学与工程学院,长沙 410082)
(2喷射沉积技术及应用湖南省重点实验室,长沙 410082)
0 引 言
日新月异的工业技术使人们的生活变得更加便利、丰富,同时也引发了能源短缺和环境污染两大世界难题。尤其是人类赖以生存和发展的水资源面临着巨大威胁,化工、化肥、造纸等工业污水源源不断地排放到河流、湖泊等水系中,造成有机染料污染水质、重金属离子含量超标等严重问题[1]。所以,治理水体污染是人类可持续发展过程中必须克服的难题之一。
目前,沉降法、物理吸附法、气浮法等纯物理技术已经广泛应用于处理工业废水,并且工艺成熟、成本低廉,但是这种非破坏式处理方法只是将污染物从液相中提取出来,并不能实现无害化,而且存在二次污染、催化剂难以重复使用等问题[2]。此外,化学生物法等降解技术也被应用于去除水中污染物,处理后可获得较好的水质,但存在成本昂贵、技术设备要求高和在氧化还原反应过程中可能产生对人体有危害的中间污染物等缺点[3]。光催化作为一种高效、清洁的技术已经成功应用在氧化降解水中有机污染物、还原重金属离子、杀灭微生物等领域。新兴的半导体材料、半导体氧化物等光催化材料具有高的光催化活性和良好的稳定性等优点,而且其在光降解水中污染物的同时也不会对环境造成二次伤害。因此这类材料在治理水体污染方面具有广阔的应用前景,引起了人们的广泛关注。
石墨相氮化碳(g-C3N4)是一种不含金属元素的新型有机半导体材料,以其独特的电子能带结构、优异的化学稳定性和热稳定性,在环境污染物降解处理、光催化选择性合成等方面显示出良好的应用前景[4]。但是g-C3N4本身是一种高激子结合能和低结晶度的半导体聚合物材料,不利于光生电子-空穴对的分离,从而致使其在光催化过程中量子效率偏低[5]。经探索发现,将g-C3N4与其他具有合适能带结构的宽禁带半导体金属氧化物进行复合,使得它们之间由于价带、导带位置和带隙能的差异而产生交迭,彼此间形成异质结构,借助g-C3N4的可见光响应性能和较高的导带位置,g-C3N4产生的光生电子可以通过紧密耦合的表面很快地注入宽禁带半导体材料的导带,从而实现光生电子-空穴对的有效分离。这种特殊异质结构的建立,可以使纳米尺度的复合光催化材料在体相内形成定向电荷通道,从而促进光生载流子的高效分离和迁移[6-10]。
本文将g-C3N4与SnO2纳米颗粒进行复合,形成具有异质结构的复合光催化剂,实现了加速光生载流子的分离、提高太阳光利用率和增强材料光催化性能的目的。
1 实验部分
1.1试 剂
实验过程中使用的试剂均为分析纯,其中三聚氰胺购于天津科密欧化学试剂有限公司,结晶四氯化锡购于上海阿拉丁生化科技有限公司,无水乙醇购于天津市致远化学试剂有限公司,罗丹明B(RhB)购于天津市恒兴化学试剂制造公司。
1.2 纯相g-C3N4的制备
以三聚氰胺为原料,采用热聚合法制备g-C3N4:称取5 g三聚氰胺粉末置于带有盖子的坩埚中,于马弗炉中520℃下煅烧4 h,所得产物研磨成粉末备用。采用上述同样的方法,通过改变煅烧温度,可以分别获得在480和560℃下煅烧4 h的产物。
1.3 g-C3N4/SnO2复合光催化材料的制备
g-C3N4/SnO2复合物由水热法制备,以50%-g-C3N4/SnO2(g-C3N4的比重为50%)为例:取1 g SnCl4·5H2O溶于40 mL水-乙醇混合溶剂(VH2O∶VC2H5OH=1∶1),加入0.43 g g-C3N4粉末,磁力搅拌30 min后超声分散30 min,得到均匀的悬浊液,然后将所得液体转移到70 mL的水热釜中,180℃保持4 h。随炉冷却后获得浅黄色沉淀物,将其用去离子水和乙醇分别洗涤3遍,离心,置于80℃烘箱中烘干,之后在马弗炉350℃煅烧1 h。通过改变g-C3N4的加入量,可以获得不同组分的复合物,分别为纯相SnO2、30%-g-C3N4/SnO2、50%-g-C3N4/SnO2、70%-g-C3N4/SnO2、90%-g-C3N4/SnO2。
1.4 仪器及表征
利用Rigaku D/Max-2500 PC型转靶全自动X射线粉末衍射仪(X-ray Diffraction,XRD)确定产品的物相和纯度,Cu Kα线(λ=0.154 2 nm)作为发射光源,加速电压为40 kV,电流为100 mA,扫描速度为5°·min-1,扫描范围:2θ=10°~80°;扫描电镜照片(FESEM)来自Hitachi S-4800冷场发射扫描电子显微镜,工作电压分别为10和15 kV;透射电镜照片(TEM)来自日本JEOL公司生产的JEM-3010型透射电子显微镜;紫外/可见吸收光谱来自Hitachi Model U-4100固相紫外/可见/近红外吸收光谱仪,扫描范围为300~800 nm,测试过程中,BaSO4作为参比材料;采用美国热电尼高力公司生产的Nicolet 5700型红外光谱仪对样品粉末进行测试,获取所得样品的红外光谱图,以溴化钾作为参照物。采用上海元析有限公司的紫外-可见分光光度计测材料的吸光度,扫描范围为300~800 nm。
1.5 光催化性能测试
通过测试所制备样品在可见光条件下催化降解罗丹明B(RhB)水溶液的能力来评估材料的光催化性能。具体的实验流程为:称取0.4 g样品粉末加入盛有400 mL RhB水溶液(10 mg·L-1)的石英杯中,搅拌均匀并置于暗处,每5 min进行取样,以6 000 r·min-1转速离心5 min后取上清液用紫外-可见分光光度计测其吸光度。反复此过程,直到RhB浓度不再变化说明吸附-脱附平衡。待吸附-脱附平衡后,开启350 W氙灯(加400 nm滤波片)对溶液进行光照,溶液与灯距离为15 cm,每30 min取一次样,离心取上清液测其吸光度。反复此过程,直到RhB染料完全降解。
2 结果与讨论
2.1 不同煅烧温度对g-C3N4产物的影响
分别在480、520和560℃对5 g三聚氰胺粉末煅烧4 h后,所得产物的产率和颜色对比如表1和图1,从中可以发现,随着煅烧温度的升高,产物产率降低,原因可能是原料在越高的温度下分解得越多,且提高煅烧温度对产物的颜色也有显著的影响。480℃煅烧后产物为浅黄色,随着温度的升高,产物颜色越来越深,最终煅烧温度为560℃时,获得土黄色产物。
对不同温度下所得产物进行了XRD表征,其表征图如图2所示。从中可以观察到520和560℃获得的产物的XRD图能与标准g-C3N4PDF卡片很好地匹配,具有分别位于2θ=27.4°和13.1°的2个主要衍射峰,分别对应于g-C3N4(002)和(100)衍射晶面,说明以三聚氰胺为原料在520至560℃温度区间内煅烧4 h均能够获得高纯度的g-C3N4[11-12]。与之相反,在480℃下获得的产物虽然也具有上述的2个主要衍射峰,但在2θ=26°左右发现一个明显的杂峰(如图中标注区域),说明在480℃下对三聚氰胺煅烧4 h后,产物中仍然存在部分聚合反应过程中生成的中间产物。综合以上结果,后续实验中所用到的g-C3N4均采用520℃下煅烧4 h获得。
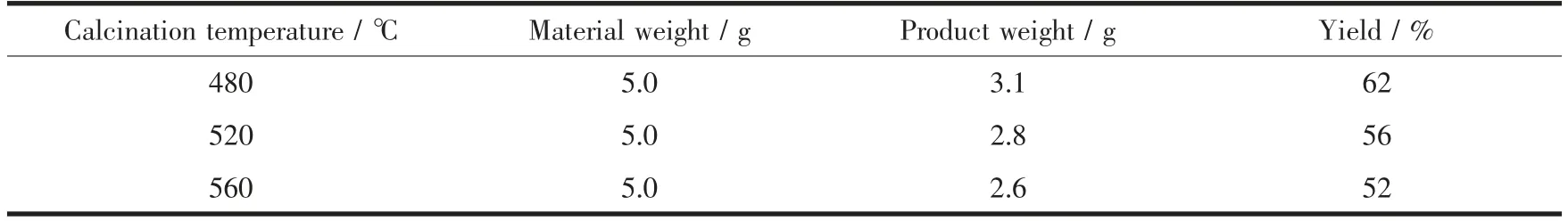
表1 不同煅烧温度获得的产物对比表Table 1 Comparison table of products obtained at different calcination temperatures

图1 不同煅烧温度获得的产物颜色比较图Fig.1 Colors of products obtained at different calcination temperatures
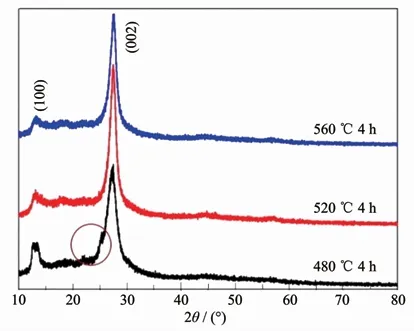
图2 不同煅烧温度所获得产物的XRD图Fig.2 XRD pattern of the products obtained at different calcination temperatures
2.2 物相结构表征
采用X射线衍射对所制备的g-C3N4/SnO2复合材料、纯相g-C3N4和纯相SnO2进行物相结构分析,所得XRD衍射图如图3所示。
在图3(a)中,纯相g-C3N4的2个主要衍射峰分别位于2θ=27.4°和13.1°,前者是环状芳香物的层间堆积特征峰,对应的晶面指数为(002),这说明样品中含有类似于石墨的层状堆积结构。后者是g-C3N4面层内嗪环结构堆垛而产生的XRD特征峰,对应的晶面指数为(100)[11-12]。对于纯相SnO2,其衍射图谱与PDF卡片(PDF No.41-1445)能很好地匹配,为四方金红石结构。其3个强峰分别位于2θ=26.6°、33.9°和51.8°,对应于(110)、(101)和(220)衍射晶面。采用谢乐公式:d=kλ/(βcosθ)计算SnO2的平均晶粒尺寸为4.1 nm。对于g-C3N4/SnO2复合材料,随着材料中SnO2含量的增加,其对应的衍射峰越来越明显,而g-C3N4的特征峰则很难从图中分辨出来,特别 是 对 于30%-g-C3N4/SnO2,50%-g-C3N4/SnO2,70%-g-C3N4/SnO2样品。因此,我们特意将2θ=20°~30°区域进行局部放大,如图3(b)。从中可以明显地看到与纯相g-C3N4和SnO2相比,复合样品在2θ=27°左右的衍射峰发生了宽化。这个衍射峰宽化可以理解为是由于g-C3N4的(002)和SnO2的(110)衍射峰发生部分重合所造成的,且随着g-C3N4含量的增加,峰宽化现象不断减弱。此外,根据XRD图中未发现有其他未知杂峰,可以判断所制备的样品纯度较高。
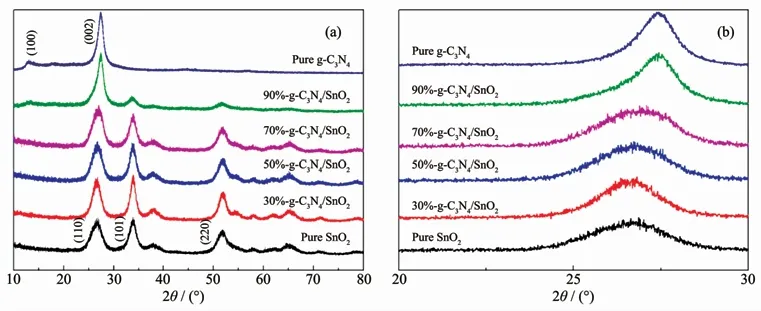
图3 (a)纯相g-C3N4、纯相SnO2和g-C3N4/SnO2复合催化剂的XRD图;(b)2θ=20°~30°区域放大图Fig.3 (a)XRD pattern of pure phase g-C3N4,pure phase SnO2 and g-C3N4/SnO2 composite catalyst;(b)2θ=20°~30°area enlargement
2.3 微观结构与形貌观察
对所制备样品的微观结构和形貌利用扫描电子显微镜(SEM)和透射电子显微镜(TEM)进行观察。如图4展示了纯相g-C3N4、纯相SnO2和50%-g-C3N4/SnO2复合催化材料的SEM图。

图4 SEM图片:(a)纯相g-C3N4;(b)纯相SnO2;(c,d)50%-g-C3N4/SnO2复合光催化剂Fig.4 SEM image:(a)pure phase g-C3N4;(b)pure phase SnO2;(c,d)50%-g-C3N4/SnO2 composite photocatalyst
从图4(a)可以观察到,纯相g-C3N4具有明显的薄层结构,并且表面非常光滑。图4(b)为纯相的SnO2,由图片可以看出,大量SnO2纳米颗粒无规则的团聚在一起且颗粒的粒径较小。对于50%-g-C3N4/SnO2样品,SnO2纳米颗粒较为均匀地分布于g-C3N4表面(如图4(c)和(d)所示),说明在高温高压的水热条件中,SnO2优先在g-C3N4表面相对粗糙的位置形核并长大,很好地防止了SnO2纳米颗粒的团聚[13]。图5为不同质量比的g-C3N4/SnO2复合材料在相同放大倍数下的微观形貌比较。图5(a)所示为30%-g-C3N4/SnO2样品,由30%g-C3N4和70%SnO2复合而成,由于SnO2的含量远超过g-C3N4,所以SnO2纳米颗粒不能像50%-g-C3N4/SnO2样品一样均匀地分散于g-C3N4的表面,而是团聚成了大颗粒。在该样品中,SnO2纳米颗粒严密地包裹着g-C3N4,使g-C3N4能接收到的光强减弱,就很有可能会对复合物的光催化活性造成负面影响。相反地,对于70%-g-C3N4/SnO2(如图5(c))和90%-g-C3N4/SnO2(如图5(d))2个样品,g-C3N4的含量较高,SnO2纳米颗粒零散地分布于g-C3N4的表面。

图5 复合物微观形貌:(a)30%-g-C3N4/SnO2;(b)50%-g-C3N4/SnO2;(c)70%-g-C3N4/SnO2;(d)90%-g-C3N4/SnO2Fig.5 Microstructure of the composite:(a)30%-g-C3N4/SnO2;(b)50%-g-C3N4/SnO2;(c)70%-g-C3N4/SnO2;(d)90%-g-C3N4/SnO2

图6 50%-g-C3N4/SnO2复合催化剂:(a)TEM照片;(b)HRTEM照片Fig.6 50%-g-C3N4/SnO2 composite catalyst:(a)TEM;(b)HRTEM
为了进一步了解复合材料的微观形貌,对50%-g-C3N4/SnO2进行TEM观察,结果如图6所示。从图6(a)可以直观地看到SnO2纳米颗粒有规则地分散于g-C3N4的表面,并且g-C3N4的层状结构清晰可辨。图6(b)为相对应的高分辨TEM图像,观察发现SnO2纳米颗粒的平均粒径为3.5~4.5 nm,与XRD的测试结果一致。图中晶格条纹间距d=0.336 nm,对应于SnO2的(110)晶面。而从HRTEM中不能观察到g-C3N4的晶格条纹,原因是g-C3N4的层内有序度较低,根据XRD的(100)衍射峰较弱可以证实[14]。
2.4 傅里叶变换红外光谱分析
采用傅里叶变换红外光谱仪对50%-g-C3N4/SnO2样品、纯相SnO2和g-C3N4进行了表征,结果如图7所示。对于纯相SnO2,位于660和535 cm-1处的2个红外吸收峰都是由SnO2中Sn-O键的伸缩振动引起的;而位于3 400和1 650 cm-1处的宽吸收峰分别对应于表面吸附水和羟基的伸缩振动[13]。对于纯相g-C3N4,可以观察到3个特征吸收带:大约位于3 200 cm-1的宽红外吸收峰是由N-H键的伸缩振动引起的[15-16];位于1 240~1 640 cm-1区域的若干个红外吸收峰都对应于C-N键和碳氮六元杂环C3N4的伸缩振动[17-18];而处在810 cm-1的红外吸收峰则对应于三均三嗪C6N7的伸缩振动[19-21]。对于50%-g-C3N4/SnO2复合样品,FT-IR图谱在810和1 240~1 640 cm-1区域都出现了g-C3N4的红外特征吸收峰,在660和535 cm-1处出现了SnO2的红外特征峰,进一步证明了复合样品中同时存在SnO2和g-C3N4两种组分。
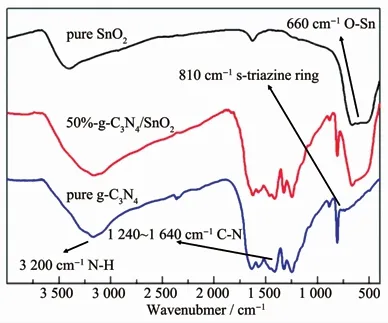
图7 纯相g-C3N4、纯相SnO2和50%-g-C3N4/SnO2复合催化剂的傅里叶红外光谱Fig.7 Fourier infrared spectroscopy of pure phase g-C3N4,pure phase SnO2 and 50%-g-C3N4/SnO2 composite catalyst
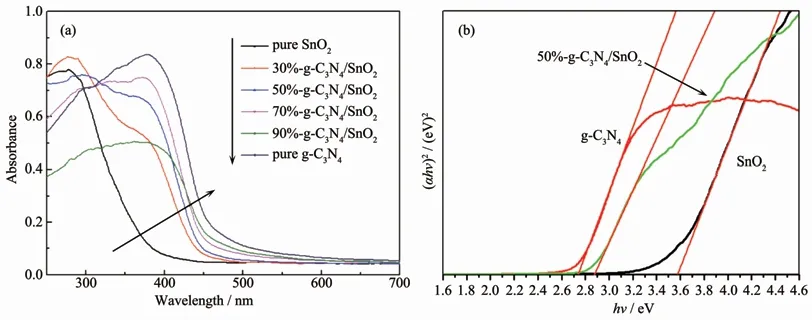
图8 (a)纯相g-C3N4、纯相SnO2和g-C3N4/SnO2复合催化剂的紫外-可见漫反射光谱;(b)(αhν)2相对于hν的变化图Fig.8 (a)UV-Visible diffuse reflectance spectra of pure phase g-C3N4,pure phase SnO2 and g-C3N4/SnO2 composite catalysts;(b)(αhν)2 vs hν
2.5 紫外-可见漫反射光谱分析
使用紫外-可见漫反射光谱仪检测纯g-C3N4、SnO2和g-C3N4/SnO2复合催化剂的吸光性能,并计算半导体材料的禁带宽度,如图8所示。在图8(a)中,纯相SnO2对光的吸收仅发生在紫外光区域,且光吸收边在380 nm左右。而纯相g-C3N4的光吸收边发生在大约460 nm处,具有较强的可见光吸收能力。此外,纯相SnO2和g-C3N4的禁带宽度可利用漫反射光谱数据根据如下公式计算获得:
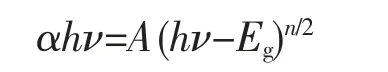
其中α、h、ν、Eg和A分别代表吸收系数、普朗克常数、光子频率、禁带宽度和常数。n的取值依赖于半导体的光跃迁类型,通常对于直接跃迁类型,n=1;对于间接跃迁类型,n=4。SnO2和g-C3N4都属于直接跃迁类型,故n值都取1。以(αhν)2为纵坐标,hν为横坐标作图,对所得曲线取切线,切线与横坐标的交点即为Eg,即所对应的半导体的禁带宽度(如图8(b)所示),计算得到的SnO2和g-C3N4的禁带宽度分别为3.59和2.73 eV。
半导体材料的导带电位和价带电位可以根据以下公式计算的到[22]:
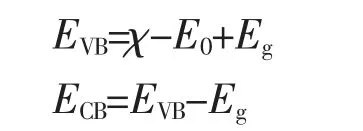
其中EVB和ECB分别代表半导体的价带电位和导带电位,E0为电子能量,标准电位下该值为4.5 eV,Eg为半导体的禁带宽度,χ为半导体的电负性,定义为各组成元素原子的绝对电负性的几何平均值(SnO2和g-C3N4分别取值为6.205和4.64 eV)[23-24]。由此,计算得到SnO2的价带和导带电位分别为+3.50和-0.09 eV;而g-C3N4的价带和导带电位分别为+1.51和-1.22 eV。对于不同组分比的g-C3N4/SnO2样品,因为g-C3N4与SnO2的紧密耦合致使复合物对可见光都有较强的吸收能力,且随着g-C3N4含量的增加,复合物的光吸收边向波长更长的方向逐步偏移(如图8(a)所示)。综合以上结果,说明g-C3N4/SnO2复合物具备了能在可见光照射下发生响应并降解有机污染物的能力。
2.6 光催化性能测试
为了评估和比较g-C3N4/SnO2复合催化材料、纯相g-C3N4和SnO2的光催化活性,我们对所有样品进行了可见光催化降解RhB水溶液测试,结果呈现于图9。图9(a)为50%-g-C3N4/SnO2样品在可见光照射下降解不同时间后的RhB水溶液的吸收光谱,从中可以看到RhB溶液的最大吸收峰处于554 nm处,但随着光照时间的延长,吸收峰的位置会逐步从554向498 nm偏移。原因是实验所制备的催化剂在光催化降解RhB分子时不是一步直接降解完全的,而是依赖于逐步脱乙基作用来完成降解。在降解过程中共有4种中间产物生成,并且最后一种中间产物为若丹明,对应的吸收峰为498 nm[25]。此外,吸收峰的强度也随着光照时间的延长快速降低,并且在180 min后几乎完全消失,说明50%-g-C3N4/SnO2催化剂对RhB染料及其中间产物都具有较强的光催化降解能力。

图9 (a)50%-g-C3N4/SnO2样品在可见光照射下降解不同时间后的RhB水溶液的吸收光谱;(b)不同催化剂在相同条件下的降解曲线Fig.9 (a)Absorption spectra of RhB aqueous solution after 50%-g-C3N4/SnO2 samples degraded under visible light for different time;(b)Degradation curves of different catalysts under the same conditions
图9 (b)为不同催化剂在相同条件下对RhB的降解曲线。对于没有加入催化剂的RhB水溶液(空白组)在同样条件下光照180 min后基本无降解,说明RhB分子在可见光照射下是非常稳定的,不会发生自降解。对于纯相g-C3N4,表现出比较差的可见光响应能力,原因可能是大量的g-C3N4片层堆积在一起且难以分离大大地抑制了它的光催化活性。此外,与纯相g-C3N4比较发现g-C3N4/SnO2催化剂样品和纯相SnO2都展现出较强的吸附能力,吸附量都超过20%。原因可以理解为水热法制备的SnO2纳米颗粒粒径较小,具有较大的比表面积,为了降低体系的表面能,SnO2纳米颗粒会在表面吸附大量的RhB分子。另外,吸附时间分别为-25和0 min时取样,测得的RhB水溶液浓度基本没有发生变化,说明了在光照前该体系已经达到了吸附-脱附平衡。在光照条件下,纯相SnO2样品对RhB溶液有一定的降解能力,而根据前文紫外-可见漫反射光谱,我们制备的SnO2样品具有3.59 eV的宽带隙,在可见光照射下并不具备降解能力。之所以能在可见光条件下降解RhB染料,是因为SnO2纳米颗粒与被吸附于其表面的RhB分子发生了染料敏化作用[26-27]。
对 于30%-g-C3N4/SnO2、50%-g-C3N4/SnO2、70%-g-C3N4/SnO2和90%-g-C3N4/SnO2复合样品,在同样条件下光照180 min后分别催化降解了62%、96%、93%和80%的RhB染料。与纯相g-C3N4相比,50%-g-C3N4/SnO2、70%-g-C3N4/SnO2和90%-g-C3N4/SnO2复合样品都表现出更优异的可见光光催化性能,并且g-C3N4∶SnO2质量比为1∶1的50%-g-C3N4/SnO2催化剂表现出了最佳的光催化活性。由此可见,复合催化材料中SnO2纳米颗粒能充分地发挥比表面积大的优势,给光生电子的转移提供了宽畅的通道,提高复合物的光催化性能。此外,g-C3N4含量的高低会直接影响g-C3N4/SnO2复合催化剂的光催化活性。当g-C3N4的含量较低时(如样品30%-g-C3N4/SnO2),过量的SnO2会将g-C3N4的表面完全覆盖住并形成保护壳,大大地削弱了照射到g-C3N4表面的可见光强度,因此表现出较弱的可见光光催化活性。同样的,当g-C3N4的含量过高时(如样品70%-g-C3N4/SnO2和90%-g-C3N4/SnO2)也会对复合催化剂的光催化性能造成负面影响。原因是g-C3N4表面过少的SnO2纳米颗粒不能满足快速分离光生电子-空穴对和转移光生电子的需求,导致电子-空穴对复合率增加。综合上述结果,g-C3N4/SnO2复合光催化剂的可见光光催化性能与g-C3N4∶SnO2质量比密切相关,两组元质量比为1∶1时(样品50%-g-C3N4/SnO2)最有利于光生电子-空穴的分离,最大程度地提高可见光的利用率。
为了更直观地比较所有样品在可见光照射下降解RhB染料的光催化性能,根据实验数据运用伪一级动力模型(公式如下)计算了实验样品降解RhB的表观速率常数[25]:
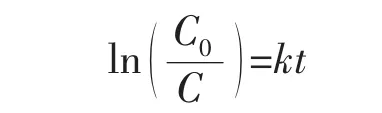
其中k为表观速率常数(min-1),C0和C分别代表RhB水溶液的初始浓度和光照时间为t时的浓度。如图10(a)所示,以ln(C0/C)为纵坐标,t为横坐标,所得的点具有直线倾向,说明本实验中光催化降解RhB染料测试符合伪一阶动力模型。计算所得的k值汇总于图10(b)中,对于纯相g-C3N4,k值为4.56×10-3min-1。对于g-C3N4/SnO2复合催化剂,随着SnO2含量的增加复合样品的光催化活性逐步增强,即k值越大。且50%-g-C3N4/SnO2复合样品的k值达到了最大值1.722×10-2min-1,是纯相g-C3N4的3.78倍。然而,当SnO2的质量比达到70%时(样品30%-g-C3N4/SnO2),相对应的k值迅速降低至4.13×10-3min-1,比纯相g-C3N4还要低。此外,对于纯相SnO2样品,由于染料敏化作用机制,对应的k值达到2.11×10-3min-1。
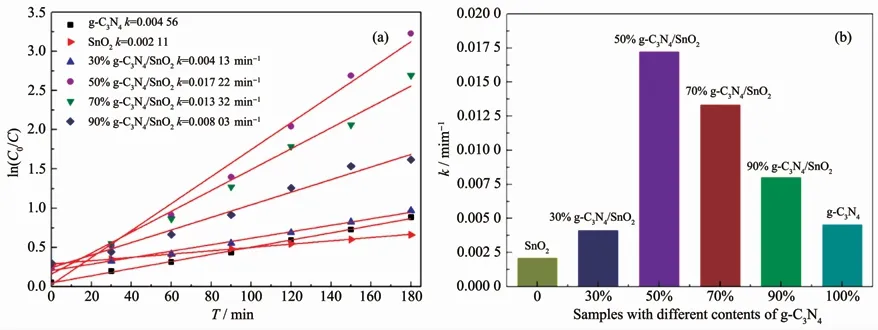
图10 (a)纯相g-C3N4、纯相SnO2及g-C3N4/SnO2复合样品在可见光条件下的光降解RhB曲线的一级动力学曲线变换图;(b)表观速率常数汇总图Fig.10 (a)First-order kinetic curve plot of photodegradation of RhB curve for pure phase g-C3N4,pure phase SnO2 and g-C3N4/SnO2 composite samples under visible light;(b)Summary of apparent rate constants
2.7 光催化机理探讨
综合上述的结果与讨论,我们提出了如图11所示的可见光催化降解水中RhB染料的作用机理。实验中g-C3N4/SnO2复合样品光催化性能提高的原因可以归结为以下2个方面:(1)g-C3N4与SnO2纳米颗粒形成的异质结在光催化降解RhB水溶液过程中发挥了重要作用,有效地促进了光生载流子的分离;(2)吸附于SnO2纳米颗粒表面的RhB分子与SnO2在可见光照射下发生染料敏化作用,加速了RhB染料的降解。根据上述实验结果,g-C3N4的导带(CB)和价带(VB)电位分别计算得-1.22和+1.51 eV,而SnO2的导带(CB)和价带(VB)电位分别处于-0.09和+3.50 eV。此外,RhB和被激发的RhB(RhB*)的氧化还原电位分别为0.95和-1.42 V(相对于标准氢电极)[28]。在可见光的照射下,处于g-C3N4价带上的电子吸收光子并获得足够能量从价带跃迁至导带,因此在g-C3N4内形成了大量的光生电子-空穴对[29-30](见方程式(1))。因为g-C3N4的导带电位(-1.22 eV)比SnO2的(-0.09 eV)更负,光生电子能通过内置电场快速地从g-C3N4表面转移至SnO2纳米颗粒。同样地,RhB染料也能受可见光激发成RhB*(方程式(2~4)),随后电子从RhB*中转移至SnO2的导带。因此,在该复合催化剂中SnO2起到了快速分离电子-空穴对的重要作用。SnO2导带上收集来的光生电子能将溶解于溶液中的O2还原成超氧阴离子(E(O2/·)=-0.046 eV vs NHE),然后进一步转变成过氧氢根(HOO·)和过氧化氢(H2O2)(方程式(5~7)),这些中间产物很快又会被继续还原成具有强氧化性的羟基自由基(·OH)(方程式(8))[31-32],·OH最终将RhB分子氧化降解成CO2和H2O等产物(方程式(9))。在整个过程中,电子的接收者(O2)控制着光生电子的转移速度[23]。留在g-C3N4表面上的空穴(h+)会直接与RhB染料反应并将其降解(方程式(10))。
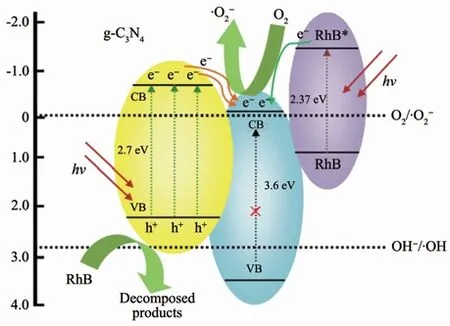
图11 g-C3N4/SnO2复合催化剂在可见光照射下催化降解RhB染料的机理图Fig.11 Mechanism of catalytic degradation of RhB dye by g-C3N4/SnO2 composite catalyst under visible light irradiation

由此可见,在本实验中RhB既是敏化剂又是被降解的污染物。当RhB*中的电子转移至SnO2导带上后,RhB分子最终变成RhB·+并很快地被其他活性物质降解掉或发生自降解[26]。考虑到吸附于SnO2表面的RhB分子并不是特别多并且RhB·+最终有可能与电子重新复合,所以,我们有理由认为,在g-C3N4/SnO2复合样品光催化降解RhB水溶液过程中,染料敏化作用的贡献是比较小的。相反地,g-C3N4与SnO2纳米颗粒之间形成的异质结构则能高效地促进光生载流子的分离,并大大地提升了g-C3N4/SnO2复合物的可见光催化性能。
3 结 论
针对g-C3N4作为半导体光催化材料使用时存在太阳能利用率偏低的问题,将其与SnO2纳米颗粒复合,形成具有异质结构的复合光催化材料。通过多种表征手段对g-C3N4/SnO2的物相结构、微观形貌和吸光特性等进行了研究,并在可见光照射条件下催化降解水中模拟污染物罗丹明B(RhB)来评估所制备样品的光催化性能,探索g-C3N4/SnO2复合光催化材料在环境污染物光催化降解方面的应用,并初步讨论了其作用机理,得出以下主要结论:
(1)采用热聚合法,以三聚氰胺为原料,在不同温度(480、520和560℃)下煅烧4 h获得3种不同g-C3N4产物。XRD结果表明,三聚氰胺粉末在520~560℃温度区间内煅烧4 h都能获得高纯度的g-C3N4,而在480℃及以下温度不能得到纯相g-C3N4。
(2)以结晶四氯化锡和三聚氰胺为初始原料,采用热聚合法和水热合成法相结合的手段,成功制备了一系列不同SnO2含量的g-C3N4/SnO2复合光催化材料。SEM和TEM结果表明,复合样品中的SnO2纳米颗粒会优先在g-C3N4表面较为粗糙的位置形核并长大,有效抑制了SnO2纳米颗粒的团聚。UVVis DRS结果表明,复合样品最大光吸收边的位置相对纯相SnO2都发生明显的红移,对光的利用能力有效增强。
(3)可见光催化降解RhB测试表明,异质结构和染料敏化的双重作用大幅度地提升了复合催化材料的光催化活性,其中最优组分(50%-g-C3N4/SnO2)的可见光催化降解RhB速率常数达到了1.722×10-2min-1,是纯相g-C3N4(4.56×10-3min-1)的3.78倍。
