四个基于酰腙配体的双核苄基锡配合物的合成、晶体结构及体外抗癌活性
2019-12-11李卓群易雨阳钟依欣余浩田谭宇星蒋伍玖
刘 骄 李卓群 易雨阳 钟依欣 余浩田 谭宇星 蒋伍玖
(衡阳师范学院化学与材料科学学院,功能金属有机材料湖南省普通高等学校重点实验室,功能金属有机化合物湖南省重点实验室,衡阳 421008)
20世纪60年代末,顺铂(Ⅱ)抗癌作用的发现及临床应用促进了金属抗癌药物的迅速发展[1],于是,设计和合成新型金属抗癌药物成为了人们的研究热点[2-4]。随着同类抗癌药物的频繁使用,多数癌细胞均会产生一定的耐药性,这就迫使科学研究人员不断地开发抗癌效果强、靶向性好、并且毒副作用小的新型抗癌药物。众所周知,二烃基锡化合物由于其优异的性质而受到人们的关注,尤其是部分化合物在体外抗癌活性上的表现优于顺铂等铂类药物[5-6]。研究表明,连接在锡原子上的有机基团及与锡原子配位的配体决定着有机锡化合物的生物活性[7-8],所以,改变其有机基团或者配体的种类可以获得具有不同活性的抗癌配合物。
酰腙类化合物本身具有一定的生物活性,分子中含有羰基氧、亚氨基氮等供电子原子,可与多种金属发生配位作用,提高其生物活性。它是一类具有易配位性、较好的热稳定性、抗肿瘤活性等特征性质而被广泛使用的金属配体[9-11]。结合本课题组前期的研究工作,我们选取含有杂环酰腙作为配体与二烃基锡配位,利用微波“一锅法”合成了4个二苄基锡配合物,并且初步测试了配合物对癌细胞的体外抑制活性,为筛选具有高抗癌活性的新型有机金属配合物奠定基础。
1 实验部分
1.1 试剂和仪器
二苄基二氯化锡和二对氯苄基二氯化锡参考文献[12]方法合成。其它试剂均为分析纯。
微波合成用意大利MILESTONE微波合成仪;IR用日本岛津Prestige-21红外光谱仪(4 000~400 cm-1,KBr压片)测定;1H、13C和119Sn NMR用Bruker AVANCE-500核磁共振仪测定;元素分析用PE-2400(Ⅱ)元素分析仪测定;晶体结构用Bruker SMART APEXⅡCCD单晶衍射仪测定;HRMS用Thermo Scientific LTQ Orbitrap XL(ESI源)测定;热重用德国NETZSCH TG 209 F3热重分析仪;熔点用北京泰克X-4双目体视显微熔点测定仪测定(温度计未经校正)。
1.2 配合物的合成
在100 mL微波反应罐中加入1 mmol 2-噻吩酰肼、1 mmol丙酮酸(或苯丙酮酸),1 mmol二苄基二氯化锡(或二对氯苄基二氯化锡),以及20 mL甲醇,在100℃下微波反应30 min,冷却到室温,过滤至锥形瓶中,控制溶剂挥发得晶体C1~C4。
配合物C1:黄色晶体,产率74.1%。m.p.108~110℃(dec)。元素分析(C23H24N2O4SSn)实测值(括号内为计算值,%):C,50.87(50.85);H,4.43(4.45);N,5.20(5.16)。IR(KBr,cm-1):3447,3103,3 080,3024,2938,2826,1614,1601,1578,1531,1491,1431,1385,1364,1 341,1 315,1 209,1 146,1 022,905,856,760,739,725,696,664,592,573,532,498,459,440。1H NMR(500 MHz,CDCl3):δ7.78(dd,J1=3.6 Hz,J2=1.2 Hz,1H),7.56(dd,J1=4.9 Hz,J2=1.2 Hz,1H),7.39~7.40(m,1H),7.13~7.15(m,1H),7.04~7.07(m,4H),6.98~7.01(m,5H),3.18(s,4H),2.06(s,3H)。13C NMR(125 MHz,CDCl3):δ170.64,153.95,136.66,131.71,131.52,128.98,128.63,128.60,128.11,127.78,125.66,30.68,13.12。119Sn NMR(Me4Sn,187 MHz,CDCl3):δ-639.29。HRMS(ESI)m/z按(C22H21N2O3SSn)+[M-CH3OH+H]+计算值:513.028 94,实测值:513.028 38。
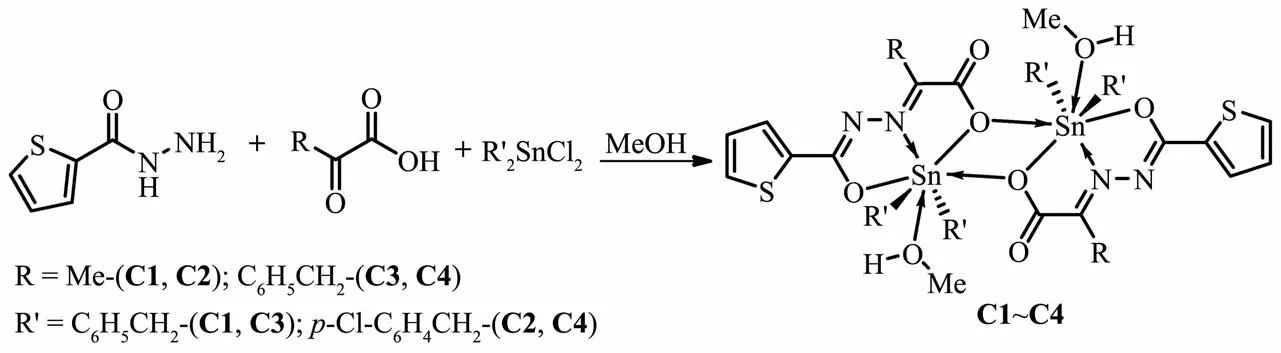
图1 配合物的合成线路图Fig.1 Synthesis of complexes
配合物C2:黄色晶体,产率73.6%。m.p.110~112℃(dec)。元素分析(C46H44Cl4N4O8S2Sn2)实测值(括号内为计算值,%):C,45.15(45.13);H,3.63(3.62);N,4.58(4.58)。IR(KBr,cm-1):3 441,3 094,3 026,2 941,1 655,1 609,1 576,1 530,1 489,1 431,1 387,1 364,1317,1209,1148,1094,1036,1015,905,858,829,739,723,650,581,536,486,469。1H NMR(500 MHz,CDCl3):δ7.80(d,J=3.6 Hz,1H),7.60(d,J=5.0 Hz,1H),7.17(m,1H),6.99(d,J=8.4 Hz,4H),6.88(d,J=8.4 Hz,4H),3.17(d,J=11.6 Hz,2H),3.12(d,J=11.6 Hz,2H),2.11(s,3H)。13CNMR(125MHz,CDCl3):δ171.15,169.37,150.34,136.63,134.82,132.26,131.93,131.42,129.48,128.36,127.82,35.74,12.80。119Sn NMR(Me4Sn,187MHz,CDCl3):δ-640.60。HRMS(ESI)m/z按(C22H19Cl2N2O3SSn)+[M-CH3OH+H]+计算值:580.950 99,实测值:580.950 56。
配合物C3:黄色晶体,产率71.3%。m.p.99~101℃(dec)。元素分析(C58H56N4O8S2Sn2)实测值(括号内为计算值,%):C,56.24(56.24);H,4.53(4.56);N,4.52(4.52)。IR(KBr,cm-1):3 433,3 076,3 022,2 938,1 634,1 612,1 599,1 576,1 530,1 491,1 481,1 452,1 431,1 385,1 356,1 342,1 312,1 227,1 167,1 128,1 032,856,758,741,696,667,579,542,497,457,440。1H NMR(500 MHz,CDCl3):δ7.79(d,J=3.6 Hz,1H),7.56(d,J=5.0 Hz,1H),7.34~7.40(m,4H),7.26~7.29(m,2H),7.19~7.23(m,2H),7.14~7.15(m,1H),6.82(s,7H),3.81(s,2H),3.19(d,J=11.7 Hz,2H),3.14(d,J=11.7 Hz,2H)。13CNMR(125 MHz,CDCl3):δ170.99,168.37,150.66,136.08,131.74,131.64,130.17,128.96,128.57,128.43,128.19,128.12,127.62,126.63,125.33,35.81,32.12。119Sn NMR(Me4Sn,187MHz,CDCl3):δ-636.27。HRMS(ESI)m/z按(C28H25N2O3SSn)+[M-CH3OH+H]+计算值:589.060 24,实测值:589.060 36。
配合物C4:黄色晶体,产率72.8%。m.p.103~105℃(dec)。元素分析(C58H52Cl4N4O8S2Sn2)实测值(括号内为计算值,%):C,50.59(50.61);H,4.83(3.81);N,4.06(4.07)。IR(KBr,cm-1):3 495,3 078,3 026,2 938,1 597,1 530,1 489,1 429,1 385,1 342,1 314,1 227,1 209,1 163,1 126,1 092,1 074,1 032,1 015,885,856,829,808,739,716,696,650,581,540,488,469。1H NMR(500 MHz,CDCl3):δ7.79(dd,J1=3.6 Hz,J2=1.2 Hz,1H),7.63(dd,J1=5.0 Hz,J2=1.2 Hz,1H),7.43(d,J=7.3 Hz,2H),7.34~7.37(m,2H),7.28(d,J=7.5 Hz,1H),7.17~7.19(m,1H),6.76(d,J=8.5 Hz,4H),6.67(d,J=8.5 Hz,4H),3.90(s,2H),3.09(d,J=11.6 Hz,2H),3.00(d,J=11.6 Hz,2H)。13CNMR(125 MHz,CDCl3):δ171.31,169.50,149.79,136.69,134.88,134.64,132.54,132.12,131.23,130.07,129.38,128.65,128.28,127.84,127.0 7,36.75,32.13。119Sn NMR(Me4Sn,187 MHz,CDCl3):δ-638.48。HRMS(ESI)m/z按(C28H22Cl2N2O3SSn)+[MCH3OH+H]+计算值:656.982 29,实测值:656.982 91。
1.3 晶体结构测定
选取尺寸分别为0.22 mm×0.22 mm×0.22 mm(C1)、0.21 mm×0.20 mm×0.20 mm(C2)、0.22 mm×0.21 mm×0.20 mm (C3)和0.20 mm×0.20 mm×0.18 mm(C4)的配合物晶体,在Bruker SMART APEXⅡCCD单晶衍射仪上,采用经石墨单色化的Mo Kα射线(λ=0.071 073 nm),以φ~ω扫描方式收集衍射数据。全部数据经Lp因子和多重扫描吸收校正。晶体结构由直接法解出,部分非氢原子坐标在随后的差值Fourier合成中陆续确定,理论加氢法给出氢原子在晶胞中的位置坐标。对非氢原子坐标及其各向异性热参数和氢原子坐标及其各向同性热参数进行全矩阵最小二乘法修正至收敛,全部结构分析计算工作采用SHELXL-97程序系统完成[13]。
CCDC:1923380,C1;1923381,C2;1923382,C3;1923383,C4。
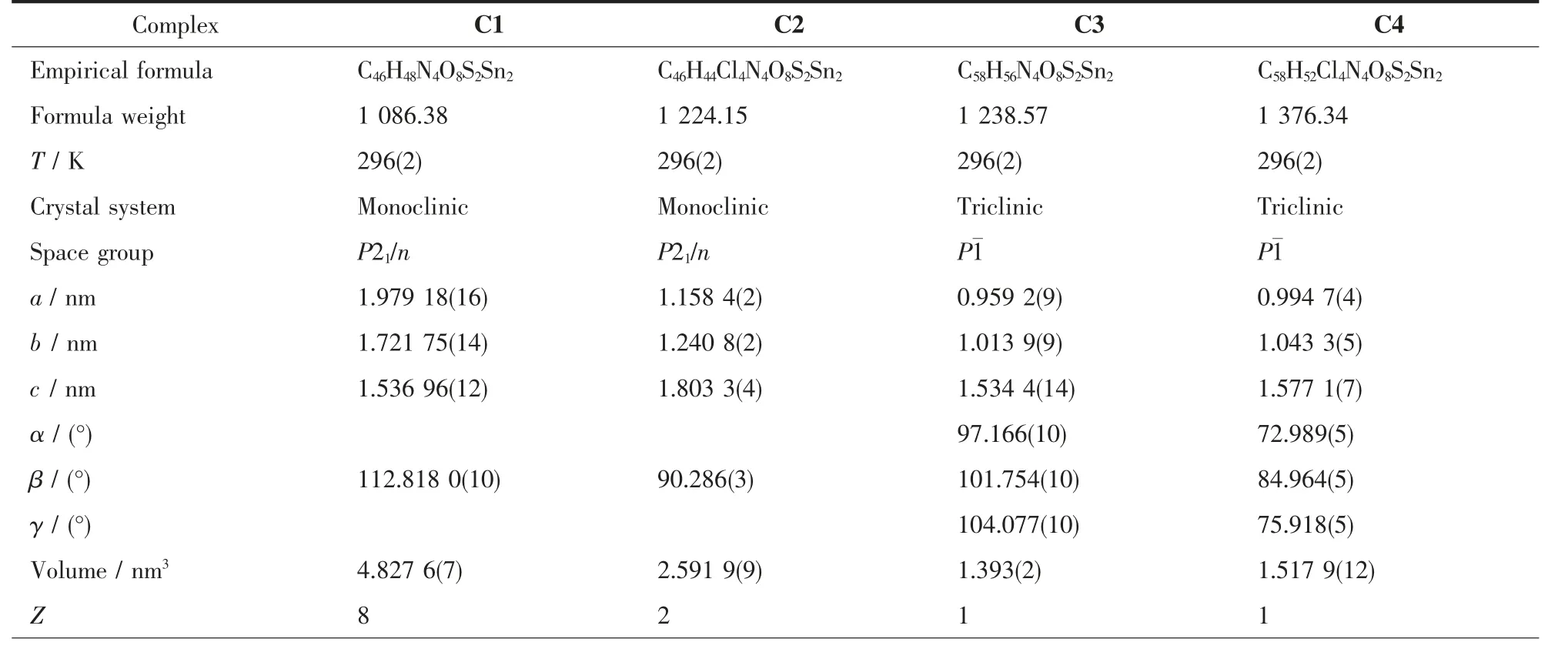
表1 配合物C1~C4的晶体学数据Table 1 Crystallographic data of complexes C1~C4
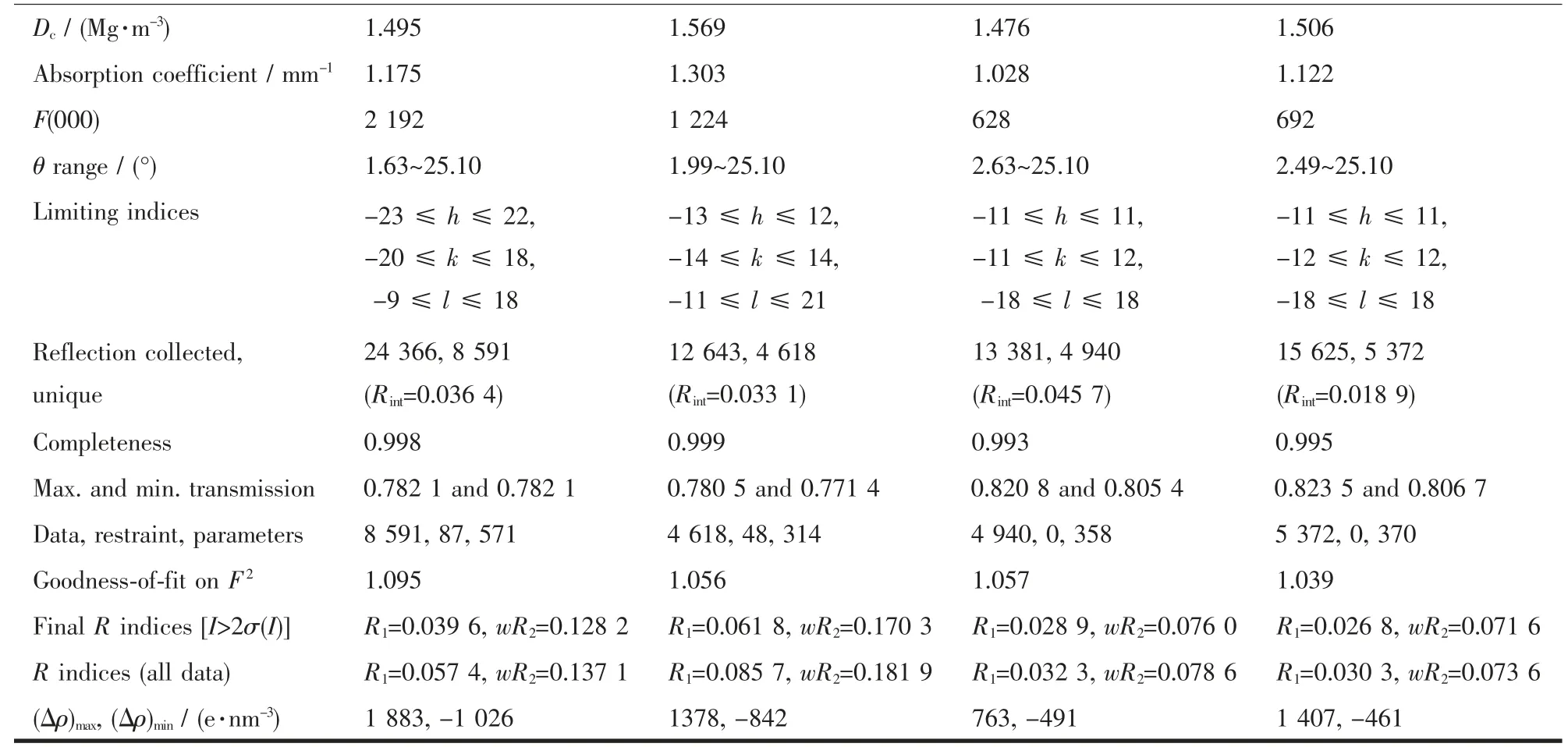
续表1
1.4 热稳定性测定
采用NETZSCH TG 209 F3热重分析仪,在空气氛下,加热速度为20℃·min-1,气体流速为20 mL·min-1的条件下,对配合物在40~800℃范围内进行了热重测试。
1.5 体外抗癌活性测试
将待测药物溶于少量DMSO,用水稀释至所需浓度,保持最终DMSO浓度小于0.1%。NCI-H460、HepG2和MCF7细胞用含10%胎牛血清的RPMI 1640(GIBICO公司)培养基,在5%(V/V)CO2、37℃和饱和湿度培养箱内进行体外培养。体外抗癌药敏试验是通过MTT法测定。数据处理使用Graph Pad Prism version 7.0程序,化合物IC50通过程序中具有S形剂量响应的非线性回归模型进行拟合得到。
2 结果与讨论
2.1合 成
合成的4个二苄基配合物C1~C4是通过2-噻吩酰肼、丙酮酸(或苯丙酮酸)、二苄基二氯化锡(或二对氯苄基二氯化锡)在微波辐射条件下“一锅法”缩合而成的。而有机锡配合物常规的合成方法有搅拌回流法、溶剂热法及界面扩散法[14],最常用的为搅拌回流法。我们在合成方法上改用微波辅助合成,相比传统加热回流方式,具有反应时间短,反应步骤简单,容易提纯,且重现性好等特点。而且4个苄基锡配合物的产率均在70%以上,对比采用传统常规方法合成的类似配合物在产率上也有了很大的提高[15]。
2.2 谱学研究
在配合物C1~C4的红外谱图中,配合物C1~C4分子中均有甲醇分子参与配位,因而在红外光谱3 433~3 495 cm-1处出现羟基的特征吸收;4个配合物在1 530~1 531 cm-1处的吸收峰归属为酰腙(C=N-N=C)键的特征吸收[16-17],C1~C4分子中羧基的反对称伸缩振动峰和对称伸缩振动峰分别在1 614和1 385、1 609和1 387、1 612和1 385、1 597和1 385 cm-1处,两者频率之差为229、222、227和212 cm-1,表明4个配合物中的羧酸根均是以单齿形式与Sn配位[18],这与X射线单晶衍射所得结果保持一致;此外,C1~C4配位键的特征峰ν(Sn-O-Sn)、ν(Sn-O)、ν(Sn-N)和ν(Sn-C)分别位于664、573、498、440 cm-1,650、581、486、469 cm-1,667、579、497、440 cm-1和650、581、488、469 cm-1处,与文献[19-20]报道的类似化合物的出峰位置一致,表明形成了4个目标配合物,并且从各个基团的出峰位置可以看出,合成的4个二苄基锡配合物具有相似的化学结构。
在1H NMR谱中,配合物各组峰的积分面积之比与预期结构的各组质子数相对吻合[21];从谱图中可以看到,配合物C1~C4中芳环上氢质子的出峰位置分别在7.78~7.01、7.80~6.88、7.79~6.82和7.79~6.67;在配合物C1和C2中,2-噻吩酰肼缩丙酮酸配体部分的甲基氢质子出峰位置分别在2.06和2.11,在配合物C3和C4中,2-噻吩酰肼缩苯丙酮酸配体部分的亚甲基氢质子出峰位置分别在3.81和3.90,其他氢原子也均具有相似的化学位移,说明这4个配合物具有相似的不对称结构单元。
在13C NMR谱中,合成的4个配合物的羧基碳、酰肼碳、亚氨基碳以及所有芳环碳均在低场出峰,配合物C1~C4中苄基上亚甲基碳原子的出峰位置分别在30.68、35.74、35.81、36.75,C1和C2中2-噻吩酰肼缩丙酮酸配体部分的甲基碳分别出峰在13.12和12.80,C3和C4中2-噻吩酰肼缩苯丙酮酸配体部分的亚甲基碳分别出峰在32.12和32.13,各组峰与理论推测结构碳原子数相吻合,这与X射线单晶衍射结果一致。
在119Sn NMR谱中,配合物C1~C4中锡原子分别在-639.29、-640.60、-636.27和-638.48处呈现1个单峰,表明4个配合物中均仅存在单一的有机锡化合物,并且4个配合物中的锡原子均在相近的化学位移处附近出峰,说明锡原子所处的化学环境相似,4个配合物具有相似的化学结构。

表2 配合物C1~C4的部分键长和键角Table 2 Selected bond lengths(nm)and bond angles(°)of complexes C1~C4
2.3 晶体结构
配合物C1~C4的主要键长和键角数据列于表2,分子结构见图2。从晶体结构图可以看出,4个新合成的配合物具有相似的不对称单元,均为双核锡分子,分子中心存在1个Sn2O2平面中心四元环,环的中心就是分子的对称中心,四元环由羧基氧原子以μ3-桥联配位Sn原子,且与2个锡原子的键长均不相等。不同的是在配合物C1分子中含有2个键参数不同的双核苄基锡,其Sn1和Sn2原子分别与另一羧基氧原子的距离Sn1-O2i为0.290 92(30)nm,Sn2-O6ii为0.280 54(33)nm(Symmetry codes:i-x,-y,1-z,ii1-x,2-y,-z for C1);而在配合物C2、C3、
C4中,Sn1-O2i分别为0.266 79(53)、0.272 28(28)、0.268 22(18)nm(Symmetry codes:i1-x,-y,1-z for C2;i1-x,2-y,1-z for C3;i1-x,1-y,-z for C4);这些数值虽大于锡原子与氧原子的共价半径之和,但是小于锡原子与氧原子范氏半径之和,它们均属于正常Sn-O键长[22],由此,说明4个配合物中的Sn-Oi键的作用均较强。

续表2

图2 配合物C1~C4的椭球率30%分子结构图Fig.2 Molecular structures of complexes C1~C4 with 30%probability ellipsoids
以配合物C1的不对称单元为例,Sn1与来自配体中的2个氧原子O1和O2,1个亚氨基氮原子N1,1个配位甲醇氧原子O4,来自2个苄基中的亚甲基碳原子C9和C16以及来自另1个配体分子中的羧基氧原子O2i等配位,形成七配位五角双锥构型。O1、O2、O4、N1、O2i占据了赤道平面的5个位置,2个亚甲基碳原子C9和C16则占据了该平面两侧的轴向位置,锡原子周围的键角O1-Sn1-N1 71.47(13)°,N1-Sn1-O2 70.78(13)°,O2-Sn1-O2i64.963(101)°,O2i-Sn1-O4 74.01(11)°,O4-Sn1-O1 78.89(13)°,均不相等;轴向C9-Sn1-C16键角为160.59(19)°,与180°偏离了19.41°,且赤道平面的5个原子与中心锡原子的键长也均不相等(Sn1-O1 0.215 8(3)nm;Sn1-O2 0.228 7(3)nm;Sn1-O4 0.243 3(4)nm;Sn1-N1 0.222 0(4)nm;Sn1-O2i0.290 92(30)nm),因此该配合物中心锡原子Sn1为七配位畸变五角双锥构型。配合物C2、C3、C4均与C1的不对称单元分子结构类似,并且键参数差异不大,中心锡原子也均为七配位畸变五角双锥构型,这种七配位的结构类型与文献[23-24]报道的配合物相似。
2.4 热稳定性研究
配合物C1~C4的热重分析曲线如图3所示。从图中可以看出,随温度的升高,4个苄基锡配合物发生相似的失重过程。由于4个配合物的中间失重阶段界限均相对模糊,所以整个失重过程分成3个阶段。在初始阶段40~180℃,配合物C1失重为5.6%(理 论 值:5.8%),C2为5.5%(理 论 值:5.2%),C3为4.7%(理论值:5.1%),C4为4.3%(理论值:4.6%),分别对应于配合物失去配位的甲醇分子;从180℃起直到配合物不再失重这一范围内,对应配合物分子失去2-噻吩酰肼缩丙酮酸配体(或2-噻吩酰肼缩苯丙酮酸配体)及苄基(或对氯苄基),4个配合物最终分 别 稳 定 在28.2%(C1)、25.1%(C2)、25.1%(C3)和21.6%(C4),残余物与SnO2的计算含量27.6%(C1)、24.5%(C2)、24.2%(C3)和21.8%(C4)基本吻合;上述热分析结果表明配合物C1~C4均在100℃之前可稳定存在,基本骨架不分解。

图3 配合物C1~C4的热重分析Fig.3 Thermogravimetric analysis curves of complexes C1~C4
2.5 体外抗癌活性研究
以卡铂作为阳性对照,应用MTT法测得配合物C1~C4对体外培养癌细胞NCI-H460(人肺癌细胞)、HepG2(人肝癌细胞)、MCF7(人乳腺癌细胞)的抑制活性,其结果如表3所示。从表中数据可知,配合物C1~C4对3种癌细胞都有一定的抑制作用,对比它们的IC50值,可以看出配合物C2、C4对3种癌细胞的抑制效果优于配合物C1、C3,并且优于对照药物卡铂。从构效关系分析,配合物中与锡原子相连的苄基对位的取代基氯原子对配合物的抗癌活性有增益作用,说明与苄基对位氯原子可能为药效基团。
在这些细胞系中,HepG2细胞系对C2最敏感,IC50值为(2.63±0.11)μmol·L-1;对于NCI-H460癌细胞,配合物C2、C4的抑制活性相当,其IC50值分别为(3.70±0.14)μmol·L-1和(3.82±0.14)μmol·L-1;对于HepG2和MCF7两种癌细胞,C2的体外抑制活性优于C4。综合上述讨论结果可知,在本文合成的4个配合物中,配合物C2相比其他配合物具有相对较好的体外抗癌活性,故其可经进一步化学优化后作为有机金属类抗癌药物的候选化合物。

?
3 结 论
利用2-噻吩酰肼、丙酮酸(或苯丙酮酸)、二苄基二氯化锡(或二对氯苄基二氯化锡)在微波辐射条件下“一锅法”合成了4个双核苄基锡配合物(C1~C4);结构分析表明,C1~C4均是以Sn2O2四元环为中心对称的双锡核分子,4个配合物的中心锡原子与配位原子均形成七配位畸变五角双锥构型。热分析结果表明,在空气氛下,配合物C1~C4均在100℃之前可稳定存在,基本骨架不分解。抗癌活性结果表明配合物C2是抑制3种癌细胞NCI-H460(人肺癌细胞)、HepG2(人肝癌细胞)、MCF7(人乳腺癌细胞)效果最好的化合物,推测其做进一步优化后可成为潜在的金属类抗癌药物的候选化合物。
Supporting information isavailable at http://www.wjhxxb.cn
