细胞焦亡在慢性脑缺血中的研究进展
2019-12-06张丽梅王恺迪综述晓审校
张丽梅,王恺迪综述,黎 晓审校
慢性脑缺血是指因各种原因所引起的慢性脑血流灌注不足导致的一组疾病。随着人口老齡化社会的进入,因慢性脑缺血导致的运动、认知、行为能力以及学习记忆等功能障碍,给患者和家人带来了严重的危害[1]。慢性脑缺血的病理生理学机制十分复杂,主要包括免疫炎性损伤、Ca2+超载、细胞自噬、细胞凋亡、氧化应激损伤、突触结构和功能异常、线粒体损伤、中枢胆碱能及单胺能系统神经递质功能紊乱等[2],其中炎症反应在慢性脑缺血方面起重要作用。脑缺血后,炎症细胞激活后引起正反馈式的炎症级联反应,将大大加重慢性脑缺血引起的神经元损伤,而脑白质和海马 CA1区的神经元损伤都将引起认知功能障碍[3]。因此,有效控制炎性反应是减轻慢性脑缺血的重要措施 。
细胞焦亡(pyroptosis)又称细胞炎性坏死,是一种由炎症性半胱天冬酶(caspase)引发的瓦解式细胞死亡形式[4]。文献表明,细胞焦亡在病毒性感染、艾滋病、动脉粥样硬化、Ⅱ型糖尿病、胃癌、白内障、酒精性肝炎、慢性婴儿神经性皮肤和关节综合征等疾病均发挥重要作用。并且有研究称,细胞焦亡还参与了慢性脑缺血的损伤过程[5~7]。故而本文拟从细胞焦亡的形态学特征、发生机制及其与慢性脑缺血的关系等角度进行综述,旨在总结细胞焦亡与慢性脑缺血之间的关系及转归中的作用,为广大学者研究和临床防治慢性脑缺血提供参考。
1 细胞焦亡
新近研究认为细胞焦亡是一种新型炎症细胞死亡类型,属于III型程序性细胞死亡的一种。根据细胞焦亡的组装生成物不同,常分为依赖caspase-1的经典炎症小体途径和依赖caspase-4.5.11的非经典炎症小体途径[8]。
1.1 细胞焦亡的形态学特征 细胞焦亡的形态学特征主要表现为,细胞发生肿胀形成凸出物、线粒体和内质网增大、质膜出现破裂并形成质膜孔,其中质膜孔的出现常常作为细胞焦亡发生的标志,此时细胞膜失去完整性,细胞渗透性增高直至细胞膜破裂,造成一些颗粒和物质(热休克蛋白、炎症因子、乳酸脱氢酶等)的释放,引起局部或全身炎症反应[9];与细胞凋亡的特征[7](凋亡小体的形成、染色质凝聚、核碎裂或核固缩、胞质DNA断裂、细胞质空泡化以及线粒体肿胀等)有着明显区别。
1.2 细胞焦亡炎症小体的组装 炎症小体(inflammasome)由Tschopp等[10]于2002年首次提出,是由胞浆内膜识别受体(pattern recognition receptors,PRRs)参与组装的一类细胞溶质内多蛋白复合物,分子量约700 KDa,它可以诱导细胞焦亡。炎症小体的组成成分主要有三种:①NOD样受体(NOD-like receptors,NLR)家族的上游传感蛋白。②衔接蛋白ASC(Apoptosi-associated speck-like protein containing a CARD),该蛋白含有CARD结构域凋亡相关斑点蛋白;该蛋白结构较为统一,由PYD-CARD表示。③下游效应子pro-caspase-1[11]。在未激活状态下,caspase带有CARD结构域,其蛋白结构式可以由CARD-Caspasedomain表示。在炎症小体组装过程下,PYD结构域和ASC的PYD结构域结合形成同型PYD:PYD相互作用,而ASC中的CARD结构域又可以和效应蛋白的CARD结构域结合形成类似的CARD:CARD相互作用,这样ASC在这里起到桥梁作用,连接受体蛋白和效应蛋白。正常情况下,pro-caspase-1不具有生物活性,但当炎性小体激活后pro-caspase-1在细胞质中可转化为异二聚体,并进一步组装成具有生物活性的四聚体caspase-1,后者可剪切pro-IL-1β和pro-IL-18,使其变成具有生物活性的IL-1β和IL-18,达到促进炎症因子成熟和分泌的作用,进而诱导细胞在炎性和应激病理条件下发生细胞焦亡[12]。
2 参与细胞焦亡的主要分子
2.1 GSDMD 经典途径中,GasderminD(GSDMD)是gasdermin蛋白家族成员之一,它被认为是caspase启动细胞焦亡的重要介质[13]。当机体受到损伤时,相关分子模式被触发:细胞膜识别受体可以检测到Toll样受体(Toll-like receptors,TLR)和 NLR,此时ASC与pro-caspase-1结合形成多蛋白复合物,caspase-1被激活进而切割GSDMD的氨基端和羧基端的连接体,产生一个N末端p30片段,p30 GSDMD在切割后结构也发生变化,可靶向作用于细胞膜并且诱导在质膜中形成大的渗透性孔,因此GSDMD被认为是形成细胞焦亡孔的候选者[14]。有学者用纯化重组的GSDMD进行体外实验发现,原子力显微镜下观察到caspase-1切割的重组GSDMD的N-末端片段紧密结合脂质体并形成直径约20 nm的圆形渗透性孔,GSDMD的N-末端片段足以导致特定形式的细胞死亡,且具有焦磷酸盐的形态学特征[15]。因此,GSDMD是细胞焦亡的直接和最终参与者。
2.2 caspase 有研究发现,caspase-1对经典途径的细胞焦亡发生起决定性作用[16]。在经典途径中,机体出现危险信号时,炎性复合体激活caspase-1,激活后的caspase-1主要作用是形成焦亡独有质膜孔,参与细胞内外离子的转运,进而导致细胞水肿而破裂[17];此外,能使IL-1β和IL-18等促炎因子募集和激活其他免疫细胞释放炎症因子、趋化因子及粘附分子等,从而放大局部和全身炎症反应[18]。此外,caspase(人caspase-4,人caspase-5和鼠caspase-11)在非经典途径中扮演了重要的角色[19]。这些蛋白质充当细胞溶质模式识别受体(PRR),当机体出现损伤(LPS刺激时),caspase-4.5.11被激活,活化的caspase-4.5.11切割GasderminD,形成氮端活性域的肽段,一方面诱导细胞膜破裂,释放内容物,另一方面诱导caspase-1的激活扩大炎症反应而加剧机体损伤[20]。因此,caspase与细胞焦亡有十分密切的联系。
3 细胞焦亡与慢性脑缺血
3.1 细胞焦亡在慢性脑缺血发生过程中的作用 炎症反应参与了慢性脑缺血,而细胞焦亡在炎症调节中起关键作用,因此,细胞焦亡与慢性脑缺血之间可能存在着密切联系[21]。在发生慢性脑缺血时,细胞通过释放损伤相关性分子模式(damage associated molecular patterns,DAMP)和病原体相关性分子模式(pathogen-associated molecular patterns,PAMP)等信号促进炎症反应,并触发了一系列复杂的分子反应,此时颅内的神经免疫炎性细胞——小胶质细胞和星形胶质细胞等出现增生和活化[22,23]。
(1)小胶质细胞的激活。一般认为,主要有经典激活(classical activation)和选择性激活(alternative activation)两种类型,其中M1型极化的小胶质细胞可释放多种炎症因子 TNF-α、IL-1、IL-6 等进一步加剧神经免疫炎性反应,使神经元损伤加重;而M2型极化的小胶质细胞被认为具有抗炎,支持和营养作用。因而研究认为,调整小胶质细胞的激活状态(极化类型)可能作为抗慢性脑缺血损伤治疗的靶点事件[24,25]。
(2)星形胶质细胞的活化。通常认为,激活后的星形胶质细胞可促使其释放释放肿瘤坏死因子、白细胞介素、生长因子等炎症因子或神经元毒性介质导致神经元的损害[26];而最新的研究表明,激活后的星形胶质细胞数量增加、胞体肥大、突触增多变粗,其标记物 GFAP 的表达增强,还可出现不同程度的折叠,损伤严重的还可形成胶质瘢痕。因此,当海马组织缺血缺氧时,海马组织的抗氧化能力将减弱,并可能释放大量炎症因子,从而加重对海马组织的损伤[27]。有研究表明[28,29],在SD大鼠局灶脑缺血后 3 h,缺血区即有IL-1β样免疫反应阳性细胞出现,主要系未活化的星形胶质细胞,直到缺血后2 m,在缺血半球仍可持续检测该阳性细胞,尤以坏死灶周围明显,且活化程度愈高。此外,还有观点认为,在caspase-1的作用下引发的细胞焦亡过程,可能通过促进IL-1β和IL-18等炎症因子的进一步释放加剧脑组织的炎性损伤,使脑缺血向更严重的程度发展[30]。
3.1.1 经典途径与慢性脑缺血 GSDMD作为细胞焦亡的执行蛋白被激活并转移到膜上后可诱导胶质细胞破裂,导致更多炎症介质的释放,从而诱导脑组织损伤和细胞死亡,这就提示经典途径可能参与了慢性脑缺血等脑组织损伤过程[31]。caspase-1蛋白酶是多蛋白炎性体复合物的核心成分,它可促进白细胞介素(interleukin,IL)-1α,IL-1β和IL-18 等大量促炎因子的释放,参与内在炎症免疫反应[32]。有研究表明[33],caspase-1低表达的小鼠可能对慢性脑缺血有一定保护作用,说明经典途径的细胞焦亡及其相关机制可能加剧了脑缺血的脑损伤。此外,用慢病毒介导的shRNA沉默caspase-1不仅显著降低了小鼠原代海马神经元中与细胞焦亡相关的蛋白质的水平,而且还降低了线粒体凋亡相关蛋白的水平[34]。此外,在大鼠的氧糖剥夺模型中,对海马切片的观察发现,抑制caspase-1可以减轻海马神经元的损伤;并且,使用caspase-1抑制剂可以减轻因不依赖于IL-1β诱导的caspase-1所导致的神经元死亡,从而神经系统损伤[35](见图1)。总之,这些研究证明了caspase-1在慢性脑缺血过程中的关键作用。因此,抑制细胞焦亡中caspase-1活性可能是治疗慢性脑缺血的一种新形式。
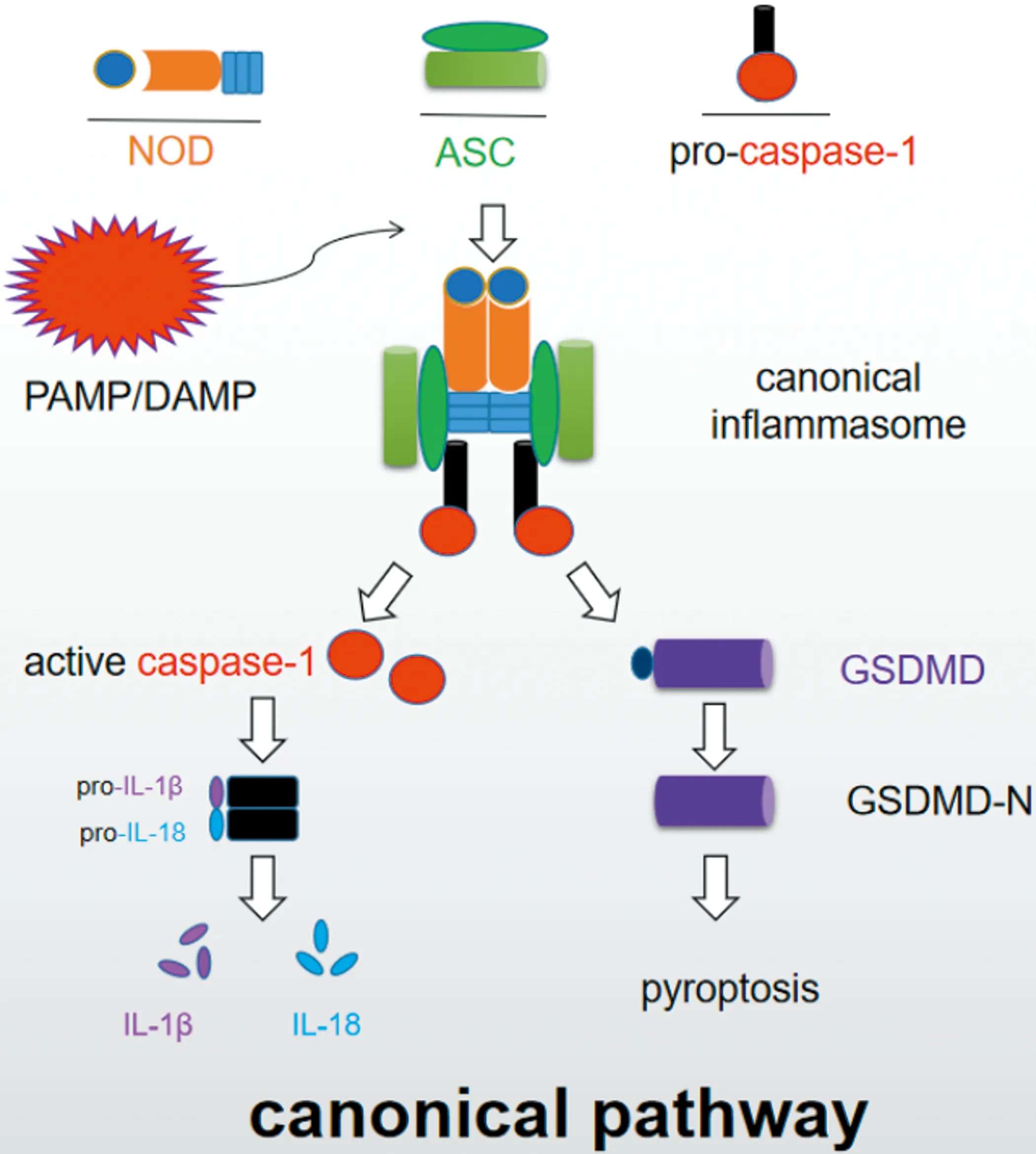
图1 PAMP/DAMP信号刺激下,NOD、ASC和pro-caspase-1装配成功为多复合物炎性小体。ASC募集酶原caspase-1,引起自动切割和激活。①激活的caspase-1可剪切没有活性的pro-IL-1β和pro-IL-18,使其变成具有生物活性的IL-1β和IL-18,促进炎症因子的成熟和分泌,诱导细胞在炎性和应激的病理条件下死亡;②通过切割GSDMD形成GSDMD的N-末端并在质膜中形成孔,诱导细胞膜破裂
3.1.2 非经典途径与慢性脑缺血 非经典途径信号分子Caspase-4参与了LPS介导的细胞死亡和NLRP3活化过程。有研究发现,在转基因小鼠中表达的caspase-4也发现具有促炎功能,但caspase-4并不能直接介导IL-1β的成熟,或者必需与caspase-1协同起作用[36],但是在钾离子的作用下NLRP3活化能使caspase-1成熟并随后释放IL-1β等炎症因子,加剧胶质细胞死亡和脑组织损伤。另一项研究表明,caspase-11的激活促进了caspase-1和caspase-3的后续激活,并且在caspase-11敲除小鼠的MCAO 模型中发现,细胞死亡的现象明显减轻[37]。caspase-11可以检测细胞内的毒素,促进IL-1α释放,caspase-11还可直接切割gasdermin D导致caspase-1依赖性IL-1β释放,从而加剧慢性脑缺血的细胞死亡[38](见图2)。因此,抑制非经典炎症小体途径可以缓和脑组织损伤作用。

图2 LPS等信号刺激下,pro-caspase-11被激活,一方面,活化的caspase-11切割GSDMD,形成GSDMD的N-末端并在质膜中形成焦亡孔,诱导细胞膜破裂,释放内容物;另一方面激活的caspase-11诱导caspase-1的激活剪切使pro-IL-1β和pro-IL-18变成IL-1β和IL-18,扩大炎症反应而加剧机体损伤。
3.2 炎症小体在慢性脑缺血损伤过程中的作用 目前已发现的炎性小体主要有四种:NLRP1、NLRP3、NLRC4和AIM2[39]。虽然大多数炎性小体具有相同的效应分子,但它们通过使用不同的受体蛋白识别不同的配体而发挥各自独特的作用。NLRP3 是研究最多且最具特征的炎性小体,属于细胞质内模式识别受体,在慢性脑缺血的发病机制中起着十分的重要作用[40],NLRP3含有PYD、NACHT、LRR三个结构域,可由LRR-NACHT-PYD:PYD-CARD:CARD-CARD-Caspasedomain表示,并对内源性损伤的各种信号作出应答,故而认为NLRP3炎症小体的激活是引起细胞焦亡的主要类型[41]。
已有研究证明,NLRP3炎症小体参与了慢性脑缺血的发生发展过程。由于NLRP3在大脑和免疫细胞中大量表达,可“感知”缺血缺氧引起的刺激信号,故激活NLRP3可促进 capase-1 前体转化为capase-1,进而促进缺血脑组织中神经元和胶质细胞的死亡,并通过IL-1β和IL-18等促炎因子的产生与释放加重神经炎症,继而影响脑梗死的预后[42~45]。另有研究发现,慢性脑缺血相关白质的损害与炎症密切相关。免疫荧光实验发现,在白质或海马中的小胶质细胞表达明显升高[46],同时,IL-1β表达增多时可以促进兴奋性氨基酸毒性和氧化毒性,进而加剧脑缺血和脑白质损害[47],这些证据表明,细胞焦亡参与了慢性脑缺血的过程[48,49]。此外,在脑缺血离体模型的研究发现,通过遗传调节抑制NLRP3后,可以有效控制神经炎症的发生,同时缓解大脑线粒体功能障碍,进而减轻缺血性脑损伤和减少神经血管并发症的发生。因此,抑制NLRP3炎症小体的形成,可为研究和治疗慢性脑缺血损伤提供新思路。
4 展 望
细胞焦亡在脑缺血中的重要性越来越受到关注。以上证据表明,细胞焦亡和炎症小体参与了慢性脑缺血,我们或许可以通过阻断或抑制炎症小体的组装、活性和(或)表达,进而抑制caspase-1(或caspase4/5/11)的激活,从而减少炎症因子的释放,达到减轻脑损伤的治疗目的。此外,我们是否也可通过阻断或抑制GSDMD的活性和表达,使细胞不形成质膜孔,阻止细胞发生焦亡,这或许是脑缺血治疗的新机制。因此,寻找各类新型caspase抑制剂和GSDMD抑制剂可能为挽救神经损伤提供新的保障,继续深入探究细胞焦亡机制和进一步认识其在慢性脑缺血进程中的作用,将会为临床治疗提供新的思路及全新的药物靶点。
