活体电化学生物传感的研究进展
2019-11-12魏欢吴菲于萍毛兰群
魏欢 吴菲 于萍 毛兰群

摘 要 化学物质参与脑内信息传递以及与脑神经相关的各种生理和病理过程,因此,脑神经化学的研究受到了越来越广泛的关注。电化学分析方法能够实现脑内重要神经分子的活体原位和活体在线分析,因而在脑神经生理病理过程的研究中具有重要意义。其中,利用酶、核酸适配体等生物识别元件,合理设计电极/溶液传感界面,进而研制出高选择性和高灵敏度的电化学生物传感器,将为脑化学的活体分析提供重要的途径。本文对电化学生物传感器在脑化学活体分析中的应用进行了评述,并对其未来的发展趋势进行了展望。
关键词 电化学生物传感器; 活体分析; 活体原位传感; 活体在线分析; 脑化学; 评述
1 引 言
作为高级神经中枢,大脑是运动、感觉、情感等生命活动的中心。因此,脑科学的研究对于理解和认识各种神经生理和病理过程的本质具有极其重要的意义。脑功能的神经信号传递绝大多数需要多种神经化学物质的共同参与,包括神经递质(如儿茶酚胺、谷氨酸、γ 氨基丁酸、乙酰胆碱、神经肽等)、神经调质(如抗坏血酸等)、能量物质(如葡萄糖、乳酸、ATP等)、离子(如H+、K+、Na+、Ca2+、Cl
等)以及其它重要的神经分子(如H2O2、H2S、NO等)[1,2]。因此,建立和发展新的分析化学的原理和方法,在活体层次实现脑化学的动态精准监测,将极大推动对脑功能和脑疾病分子机制的研究。
目前,用于活体层次的分析和成像技术主要包括电化学方法、荧光成像[3]、质谱成像(Imaging mass spectrometry, IMS)[4]、磁共振成像(Magnetic resonance imaging, MRI) [5]、磁共振脑功能成像(Functional magnetic resonance imaging, fMRI)[6]、磁共振波谱(Magnetic resonance spectroscopy, MRS)[7]、正电子发射断层扫描(Positron emission tomography, PET)[8]和单光子发射断层扫描(Single photon emission computed tomography, SPECT)[9]等。與电化学分析方法相比,这些成像技术具有无损的优势,但是它们常需使用一些特殊的化学试剂作为探针,可分析的神经分子种类较少,且时空分辨率较低。电化学分析方法因其具有高时空分辨、可实现活体、原位、实时以及多组分同时分析等优点,在神经分析化学的研究中备受关注[10~22],已被成功应用于脑内部分重要神经分子(如多巴胺、抗坏血酸等)的活体分析[23~33]。
生物传感器诞生于1962年,Clark等[34]率先提出了酶传感器的概念。自第一支酶电极问世以来,随着物理、化学原理及方法的不断引入,生物传感器的研究已发展成为一个多学科高度交叉与融合的前沿领域之一[35]。电化学生物传感器通过对电极表面进行功能化修饰的基础上,识别元件将待测分子转化为电化学可检测的物质,从而实现物质浓度的选择性定量分析[36,37]。按生物识别元件分类,电化学生物传感器可分为酶传感器、免疫传感器、DNA传感器、微生物传感器等。由于其高的选择性和表界面设计的多样性,生物电化学传感器在脑化学活体分析中展示出独特的优势,其中酶传感器由于特异性高、响应时间短,特别适用于活体分析。本文针对脑内电化学活性相对较差的分子,如能量物质(葡萄糖、乳酸、ATP)、神经递质(多巴胺,谷氨酸、乙酰胆碱)等,按照不同的识别机理,分别对基于氧化酶、脱氢酶、漆酶、谷氨酸合成酶、核酸适配体(Aptamer)及多酶协同的电化学生物传感器在活体分析中的应用进行评述,并对其未来的发展趋势进行了展望。
2 基于氧化酶电化学生物传感器的活体分析
2.1 第一代氧化酶型生物传感器
第一代氧化酶型生物传感器是利用O2作为氧化酶的电子受体,通过检测酶催化反应过程中H2O2的生成量,进而实现被测物浓度及其变化的传感分析。尽管目前大部分氧化酶型生物传感器是基于该原理研制而成,但是该类生物传感器仍面临诸多问题[38]。一方面,O2作为酶催化反应的电子受体,其浓度随环境的波动将会影响传感器信号的稳定性; 另一方面,H2O2的电化学氧化通常具有较高的过电位,而脑内共存的其它物种,如多巴胺及其代谢产物、抗坏血酸等,在此高电位下也能发生电化学氧化反应,进而干扰测定; 虽然检测H2O2的还原电流能够避免以上物质氧化的干扰,但由溶解氧电化学还原而产生的干扰仍是一个不可回避的问题[39]。
为了提高第一代氧化酶型生物传感器的选择性,研究人员曾在传感器的表面再覆盖一层离子交换膜[40]或者电化学聚合膜[41],从而抑制电化学活性物质(如抗坏血酸)向电极表面的扩散和的电化学氧化。抗坏血酸对于传感器的干扰也可通过在电极表面或在线电化学传感器上游引入抗坏血酸氧化酶修饰层或酶柱,预先氧化抗坏血酸进而消耗其含量实现[42]。Baker等[43]在铂微电极表面电聚合邻苯二胺薄膜,并修饰以甲基丙烯酸甲酯、醋酸纤维素等作为稳定剂,结合生物识别元件(胆碱氧化酶)实现了大鼠脑内胆碱的原位电化学分析。Li等[44]通过在葡萄糖氧化酶修饰的电极上电聚合一层邻苯二胺薄膜,提高了对葡萄糖的选择性,并将该阵列电极成功用于大鼠扩散性抑制(Spreading depression,SD)过程中葡萄糖、O2和电生理活动的同时测定。他们发现,在SD过程中,脑内葡萄糖和氧分压会发生明显的变化。Chatard等[45]利用气相沉积的方法在直径7 μm的碳纤维表面镀铂,再电聚合一层间苯二胺薄膜,较好地抑制了内源性电活性分子向电极表面的扩散。通过使用葡萄糖氧化酶和乳酸氧化酶,他们研制出了对脑组织创伤较小,但对于葡萄糖和乳酸具有良好响应的活体电化学生物传感器,成功用于脑神经生理病理模型中葡萄糖和乳酸动态变化的研究。他们还发现,在SD过程中,传统微电极和碳纤维微电极对葡萄糖和乳酸的响应表现出较大差异。
除上述方法外,背景扣除的方法也能消除干扰。Gerhardt研究组[46~48]在阵列电极上设计自参照电极,将其电流信号作为背景信号,在具体的分析测定中予以扣除,这种方法可消除在相同的极化电位下其它物质对谷氨酸氧化酶修饰电极的干扰。他们首先在电极表面修饰一层Nafion,避免抗坏血酸的干扰; 随后,利用戊二醛和牛血清白蛋白(Bovine serum albumin,BSA)交联法将谷氨酸氧化酶固定至阵列电极表面,用于记录氧化电流的总和; 相邻的自参照位点仅修饰BSA和戊二醛,用于记录背景氧化电流。二者电流之差用于谷氨酸的定量分析(图1A)。他们利用局部注射谷氨酸的模型,成功地将该生物传感器用于鼠脑谷氨酸活体原位的实时监测,并实现了自由活动大鼠在静息状态及应激压力下脑内谷氨酸的长期监测(图1B)。
在活体电化学分析中,除利用微电极技术而发展的活体原位电化学分析方法外,还有一类是基于微透析取样技术的活体分析方法。电化学生物传感器不仅可实现神经分子的活体原位传感分析,而且也可作为高选择性的检测器,通过结合微透析取样技术,实现脑化学的活体在线分析。
微透析取样技术自1972年问世以来,已被广泛应用于神经科学、药学和分析化学等多学科的研究中[49]。作为活体取样技术,该技术一般需要结合样品分离和检测,方可实现与脑化学相关的研究。电化学生物传感器由于具有高选择性和传感界面设计多样性等优点,因此微透析技术和高选择性生物电化学传感的有效结合,可形成活体在线电化学分析系统(Online electrochemical system,OECS),实现部分神经分子(如葡萄糖、乳酸、谷氨酸等)的直接检测[50]。相对于使用样品分离的离线分离分析,OECS具有时间分辨率高、样品保真、易与行为学研究相结合等优点[51]。但是,无需样品分离的直接检测方法要求在线电化学传感器应满足以下条件:(1)高选择性:应避免脑透析液中其它神经分子,如抗坏血酸、尿酸、多巴胺及其代谢物的干扰; (2)高灵敏度:可有效检测脑透析液中的低浓度物质,如多巴胺、谷氨酸、乙酰胆碱等; (3)良好的稳定性和重现性:可进行长时程的流动分析; (4)多组分同时分析:多个传感器之间应无交叉干扰; (5)与生理学研究的兼容性:能够实现在复杂脑神经生理和病理条件下对于特定神经分子的专一性连续检测[52]。
基于氧化酶构建的第一代电化学生物传感器已被用于构建活体在线分析系统。Rogers等[53]通过电聚合苯酚将葡萄糖氧化酶固定在微流控芯片的工作电极上,有效避免了抗坏血酸、多巴胺等内源性电活性分子的干扰。他们结合快速微透析取样技术,研究了SD过程中脑内葡萄糖的变化规律; 通过使用地塞米松抗炎药物,实现了脑内葡萄糖浓度的长期监测[54]。为了解决H2O2检测时的过电位问题,辣根过氧化物酶(Horseradish peroxidase,HRP)作为H2O2还原的高效生物酶催化剂,常被用于构筑高选择性的电化学生物传感器。在该类传感体系中,通常需要外加电子媒介体实现HRP和电极之间的电子传递。Niwa等[55]制备了固定有谷氨酸氧化酶的反应器,将锇的配合物与HRP复合而形成的凝胶(Os gel HRP)修飾在玻碳电极上,并以此作为检测器,发展了谷氨酸的在线分析方法,灵敏度高(24.3 nA/(μmol/L)),检出限低(7.2 nmol/L),成功检测到KCl刺激单个神经元细胞及电刺激脑切片引起的亚微摩尔及微摩尔水平的谷氨酸变化。Osborone等[56]制备了一种双半圆形的工作电极,分别在两个半圆电极上修饰了Os gel HRP/葡萄糖氧化酶和Os gel HRP/乳酸氧化酶,在克服了电极间交叉干扰的前提下,建立了脑内葡萄糖和乳酸浓度同时检测的活体在线电化学分析方法,实现了大鼠在清醒状态下大脑纹状体中葡萄糖和乳酸的连续监测(图2)。
Mao等[57]将Os gel HRP和次黄嘌呤氧化酶同时固定在电极上,建立了检测次黄嘌呤的在线电化学分析方法,将检测电压置于200 mV, 避免了抗坏血酸等物质的干扰,也提高了检测的灵敏度。此外,他们将利用氧化酶构建活体在线分析方法的传统思路拓展,提出利用其它分子作为HRP电子传递媒介体发展活体在线分析方法的新策略[58],通过在电极表面电聚合麦尔多拉蓝(Meldola's blue,MB),实现了HRP与电极之间的界面电子转移,建立了葡萄糖和胆碱测定的在线电化学分析方法。
虽然利用HRP可相对选择性地测定H2O2,但易受到内源性抗坏血酸的干扰。抗坏血酸干扰H2O2的测定可通过以下4个途径:(1)抗坏血酸在电极表面的直接电化学氧化; (2)抗坏血酸在金属离子(如Fe3+或Cu2+)的催化下与H2O2反应; (3)HRP催化抗坏血酸与H2O2的化学反应; (4)抗坏血酸和HRP的部分媒介体(如Os gel HRP)发生化学反应(图3B)。针对此问题,Mao等[59]利用环盘电极的结构特点,在中心的盘状电极上修饰抗坏血酸氧化酶,实现了脑透析液中抗坏血酸的预氧化; 在环状电极上,利用共沉积的方式修饰HRP和媒介体聚吡咯(polypyrrole,PPy),避免了抗坏血酸和媒介体之间的反应,同时实现了H2O2的选择性检测。此外,他们在HRP/PPy电极外层再修饰聚苯酚膜(polyphenol,PPh),提高电极的选择性。在进行活体在线分析时,为了抑制抗坏血酸和H2O2在以金属离子为催化剂时可能发生的化学反应,他们在在线体系中引入含有EDTA的缓冲溶液,一方面稳定了检测系统的pH值和氧分压,另一方面EDTA螯合了金属离子,进而成功地实现了脑透析液中H2O2的活体在线电化学分析,如图3A和3C所示。
针对“天然酶”HRP不稳定性的问题,Lin等[60]使用“人工模拟酶”普鲁士蓝(PB)代替HRP,实现了对H2O2的选择性传感, 结合氧化酶,实现了葡萄糖和乳酸的活体在线电化学分析(图4)。
Ma等[73]以沸石咪唑酯框架(Zeolitic imidazolate frameworks,ZIFs)材料为载体,利用其多孔性、高比表面积、结构和功能可调等特性,实现了电化学催化剂(MG)和葡萄糖脱氢酶的共固定,以及NAD+在复合层中的快速传输。活体分析结果表明,利用ZIFs实现的MG和葡萄糖脱氢酶的共固定的研究思路,不仅为脑神经电化学分析提供了新策略,也为新型传感器的设计和构筑提供了新途径[74]。Huang等[75]利用辅酶NAD+在室温下可与金属离子Tb3+形成一种无限配位聚合物(Infinite coordination polymer,ICP)的性质,在配位过程中引入生物识别单元(葡萄糖脱氢酶)和MG,发展了“一锅法”制备同时包含所有传感元件(酶、辅酶因子、电催化剂)的纳米结构, 该纳米结构具有优良的生物电化学活性,可方便地修饰于电极表面。然而,ICP纳米颗粒本身不具备导电能力,因此,所制备的电化学生物传感器灵敏度较低。针对此问题,Lu等[76]將这种ICP纳米颗粒与SWNTs进行复合,制备了均匀分散的ICP/SWNTs复合物,可通过简单的滴涂法在电极表面形成三维导电网状结构,大大提高传感器的灵敏度和稳定性。该研究不仅为脑神经化学的活体分析提供了新方法,也为脱氢酶电化学传感器中传感元件在电极表面一体化固定提供了新策略。
4 基于漆酶电化学生物传感器的活体分析
漆酶是一种蓝铜族氧化酶,可催化酚类物质的氧化和O2的还原[77]。该酶含有的4个Cu2+位于蛋白的疏水空腔内,其中T1 Cu2+距蛋白表面约0.6 nm,是催化过程中接收外来电子的第一站,而由T2和T3 Cu2+形成的三核铜簇是O2的结合位点,经由蛋白内电子传递途径接收来自T1 Cu2+的电子,从而将O2还原成H2O。目前,漆酶已被广泛应用于生物燃料电池、生物传感、废水处理等领域。漆酶独特的性质也为活体分析化学提供了新途径。多巴胺是一种儿茶酚胺类递质,参与神经信号的传递,在奖赏、运动、成瘾等过程中发挥无可替代的作用[78]。多巴胺本身具有邻苯二酚的结构,也是漆酶的底物之一。基于多巴胺在电极表面发生电化学 化学 电化学反应的机理,Xiang等[79]率先提出通过测定多巴胺氧化产物5,6 二羟基吲哚啉醌的还原电流,进而间接测定多巴胺的新思想。他们利用漆酶催化多巴胺的第一步氧化反应,继而驱动后续反应的发生; 其最终产物5,6 二羟基吲哚啉醌具有较好的电化学活性,在较低电位下可发生电化学还原。同时,抗坏血酸和多巴胺代谢产物3,4 二羟基苯乙酸也可被漆酶催化氧化,生成非电化学活性的物质,从而实现了在不受抗坏血酸等氧化电流干扰的情况下对多巴胺的间接测定。随后,Lin等[80]合成磁性颗粒,并通过共价作用将其表面进行漆酶功能化。在磁场作用下,将磁性颗粒填充于石英毛细管内壁,形成磁性漆酶微反应器,并将其置于在线电化学检测器上游,从而建立了多巴胺的活体在线分析方法(图7)。
从基础生物电化学的角度,当漆酶的疏水腔朝向电极表面时,T1 Cu2+易与电极之间实现有效的直接电子转移,但是调控漆酶在电极表面的朝向一直是生物电化学研究领域最关注且富有挑战性的问题之一[81]。2006年,Zheng等[82]利用碳纳米管修饰电极实现了漆酶与电极之间的直接电子转移,该研究为漆酶直接电化学的研究提供了新的策略。为了提高生物电化学催化的电流密度,Wu等[83]通过在漆酶溶液中加入20%乙醇,有效地提高了漆酶与碳纳米管之间的相互作用,使得漆酶在碳纳米管上形成了有利于直接电子转移的分子朝向,从而将O2的催化还原电流提高了6倍。随后,Han等[84]探究了不同碳材料(碳纳米管、碳球、石墨烯)用于电极表面固定小漆酶(Small laccase, SLAC)时对于O2还原催化效率的差异,发现SWNTs SLAC修饰的电极对于O2还原的催化电流最大。基于此,他们提出了纳米碳材料表面曲度调控小漆酶直接电催化行为的模型,为基于纳米碳材料的生物电化学界面设计与构筑提供了新的方法。
近期,Wu等[85]利用漆酶能在相对高的电位下催化氧还原特性,使用漆酶修饰的碳纤维电极作为指示阴极,并利用内充有弱酸性缓冲液的玻璃毛细管作为其工作环境,从而稳定指示电极电位; 同时,以神经调质抗坏血酸为检测分子,使用碳纳米管修饰的碳纤维电极为阳极,构建了以氧化还原电势为信号读出方式的电位测定方法(Galvanic redox potentiometry,GRP),如图8所示。该原理的提出使得电化学信号和电生理信号的同步记录成为可能,为研究脑神经活动中化学信号和神经电活动之间的关联提供了新方法。
5 基于谷氨酸合成酶的生物传感
除了氧化酶和脱氢酶作为生物识别元件被广泛应用于电化学生物传感领域之外,自然界还存在着种类繁多的其它酶类,如固氮酶、氢化酶等,它们被应用于能源转换、电催化以及电合成等领域。针对目前基于氧化酶和脱氢酶的电化学传感器面临的一系列问题,基于其它酶类的活体电化学生物传感原理的设计和构筑显得尤为重要。谷氨酸合成酶是固氮过程中实现氨同化反应的关键酶,仅存在于微生物和高等植物部分组织中,并参与相应的氨基酸代谢和光转换等过程。目前,谷氨酸合成酶的晶体结构已被解析,但其在电催化领域的研究尚未被报道。2018年,Wu等[86]首次将蓝藻细菌中的铁氧化还原蛋白和以铁氧化蛋白为电子供体的谷氨酸合成酶在大肠杆菌体内完成重组和表达。该谷氨酸合成酶主要由氨基转移酶中心、黄素单核苷酸(Flavin mononucleotide,FMN)和铁 硫结合中心组成。他们利用具有不同氧化还原电位的电子传递媒介体合理构筑电化学界面,实现了催化谷氨酸的正向合成和反向氧化两个反应过程,如图9所示。该酶在催化谷氨酸氧化过程中不受O2干扰,也无需外加辅酶,为设计谷氨酸检测的活体电化学生物传感器提供了新思路。
6 基于核酸适配体(Aptamer)的生物传感
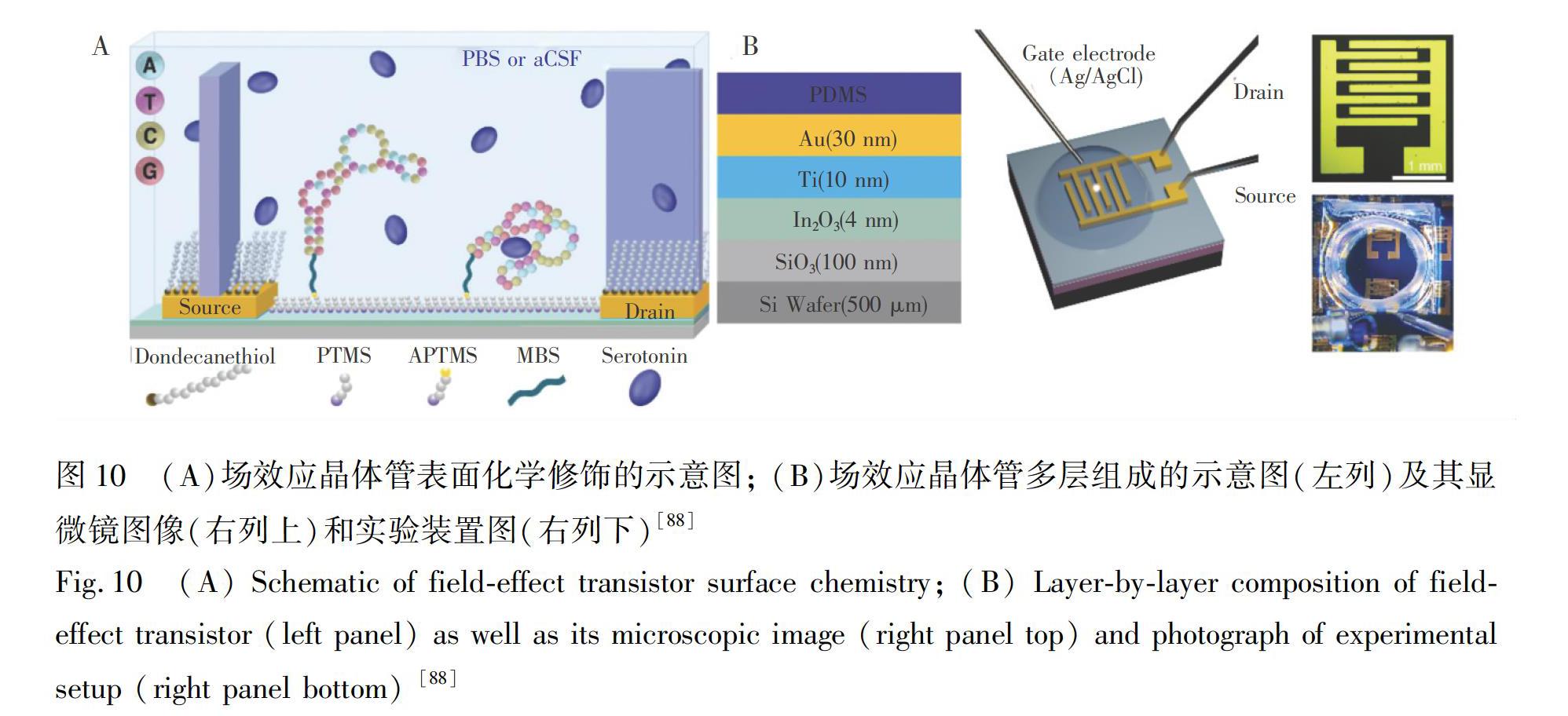
目前,Aptamer已成为诊断和治疗的重要分子工具。与天然受体(如抗体和酶)相比,Aptamer作为生物识别元件,在电化学生物传感领域具有很好的优势:(1)针对具体的靶标(从小分子到尺寸较大的蛋白质甚至细胞),理论上都可通过体外筛选的方法,得到具有高特异性和亲和力的Aptamer; (2)Aptamer具有化学稳定性; (3)Aptamer在与靶标结合时常能发生显著的构象变化,该特点可为高灵敏度和高选择性活体电化学生物传感原理的设计和构筑提供可能[87]。2018年,Nakatsuka等[88]在超薄金属氧化物场效应晶体管阵列上修饰能够特异性结合靶标的Aptamer,在生理条件下,实现了5 羟色胺、多巴胺、葡萄糖、1 磷酸神经鞘氨醇的选择性检测,如图10所示。靶标分子和Aptamer结合诱导后者带负电的磷酸二酯骨架发生构象变化,引起栅极调控半导体通道导电能力的改变,进而实现了待测靶标的高灵敏检测。
基于Aptamer的ATP生物传感器多见报道,然而,目前广泛使用的Aptamer亦可与ADP和AMP结合,对于ATP的识别专一性较差,因此限制了该类传感器在复杂体系(如活体)分析中的应用。为了提高对ATP检测的选择性,Yu等[89]报道了一种具有双识别单元的高灵敏、高选择的ATP传感器。该传感器巧妙地结合了Aptamer对A碱基的识别能力和基于咪唑的阳离子聚合物Pim对三磷酸根的强结合能力,有效提高了对ATP识别的专一性,实现了脑透析液中ATP的高选择性活体分析。
7 多酶协同电化学生物传感器的活体分析
神经系统中存在着一类重要化学物质,如乙酰胆碱[90]、ATP[91]、γ 氨基丁酸[92]等,既没有电化学活性,也缺少相对应的氧化酶或脱氢酶识别元件,利用一般的电化学生物传感原理很难实现其直接检测。因此,多酶串联反应的电化学生物传感器应运而生。如乙酰胆碱的传感分析可同时利用乙酰胆碱酯酶(Acetylcholine oxidase, AChE)和胆碱氧化酶(Choline, ChOx),通过检测酶促反应过程中H2O2的生成量实现乙酰胆碱的传感分析。但是,在中枢神經系统中,除抗坏血酸、尿酸等具有电化学活性的物质外,细胞间液中胆碱的浓度比乙酰胆碱的浓度一般高1000倍左右,这些物质都会干扰乙酰胆碱的测定。因此,消除胆碱和抗坏血酸的干扰是乙酰胆碱活体分析的关键。Niwa等[93]在微透析取样 在线电化学检测系统研制的过程中,在乙酰胆碱电化学传感器的上游引入固定有胆碱氧化酶和过氧化氢酶(Catalase)的微柱,先将胆碱及其氧化产生的H2O2消耗掉,从而避免胆碱对乙酰胆碱的干扰。为了降低抗坏血酸的干扰,他们在AChE ChOx/Os gel HRP修饰层上修饰了Nafion膜,阻止带负电荷的抗坏血酸向电极表面扩散。基于此,他们建立了乙酰胆碱的高选择在线电化学分析方法,在大鼠海马脑切片上,成功检测到电刺激诱导的胞外乙酰胆碱浓度的升高。
Burmeister等[94]设计了一种多位点的微电极阵列,实现了脑内胆碱和乙酰胆碱的同时原位测定。为了排除抗坏血酸和多巴胺的干扰,首先在铂电极表面电聚合一层间苯二胺膜。两个铂记录位点只修饰胆碱氧化酶,用于获取胆碱的浓度信息; 另两个位点同时修饰乙酰胆碱酯酶和胆碱氧化酶,其电流信号与前者的差值即可用于乙酰胆碱的定量分析。
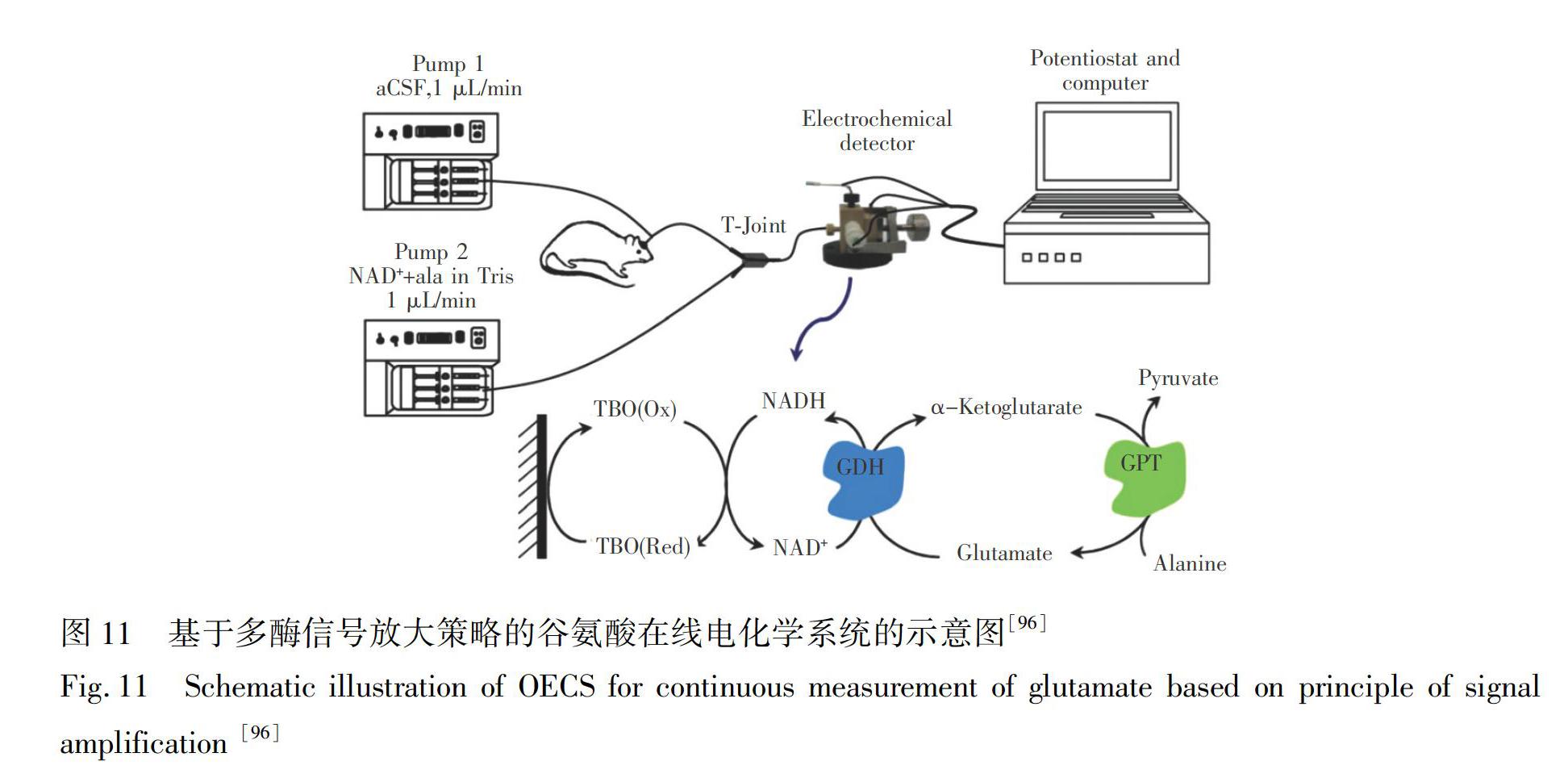
除此之外,针对中枢神经系统中部分有相对应氧化酶或脱氢酶识别元件,但生理浓度较低的神经化学分子(如谷氨酸[95]),为了满足活体检测的需求,该类电化学生物传感器的设计常采取多酶信号放大的策略,以实现底物的循环,提高检测灵敏度。Zhang等[96]利用谷氨酸脱氢酶为识别元件,谷丙转氨酶实现谷氨酸的循环,在进行活体在线分析时,在在线体系中加入丙氨酸启动谷丙转氨酶的酶促反应,实现谷氨酸测定信号的循环放大,进而成功实现了脑透析液中谷氨酸的活体在线电化学分析(图11)。
多酶协同联用的电化学生物传感器也被拓展用于腺苷和ATP的活体分析。腺苷是一种能够调节心率、睡眠和呼吸的神经调质; ATP则是生物体内最直接的能量来源,同时也参与多种信号的转导。Llaudet等[97]在铂微电极上修饰黄嘌呤氧化酶、嘌呤核苷磷酸化酶和腺苷脱氨酶三种酶,通过酶促反应将腺苷转化为电化学可检测的H2O2。基于这种设计模式,他们发展了一种尺寸小(25~100 μm)、响应快((2±0.23) s)、灵敏度高(100~222 mA/(mol/L cm2))的腺苷电化学生物传感器,并将其应用于缺氧状态下海马体切片腺苷释放的活体分析。他们还通过在铂微电极表面修饰含有甘油激酶和甘油 3 磷酸氧化酶的薄层,发展了一种针对ATP检测的多酶型电化学生物传感器[98]。该传感器响应时间短(10%~90%电流响应时间<10 s),灵敏度高(~250 mA/(mol/L cm2))。他们利用该电化学生物传感器,首次发现了ATP在中枢神经系统化学感应转导过程中的重要作用[99]。
8 总结与展望
目前,基于电化学生物传感器的活体分析已经成为分析化学、神经科学、物理和材料科学等多学科交叉研究领域的热点之一,对于推动脑神经生理和病理分子机制的研究具有重要意义。本文综述了多种电极/溶液界面的设计策略,旨在建立和发展可用于活体脑化学分析的生物电化学传感器。然而,在活体层次上准确地破译化学信号和大脑机能之间的关系仍然面临着巨大的挑战,生命体系的复杂性,以及分子间相互作用的多样性,对神经化学物质的活体测定提出了更高的要求。创新发展生物传感的原理,并研制新型活体可用的传感器,进而开展全新的神经化学研究,将是未来活体生物传感器研究的核心内容之一。首先,随着电化学原理和方法的发展,发展新的传感原理,如基于GRP[85]、离子传输[100]、有机电化学晶体管[101]等原理的电化学生物传感器,将为神经化学分析研究提供新的思路。其次,随着新型材料的不断涌入,如石墨炔[102]、单原子催化剂[103]、钙钛矿[104]等,电化学生物传感器的研究将迎来新的机遇。另外,具有良好导电性和生物兼容性的柔性材料的出现,将为柔性微型传感器的设计提供可能。利用柔性电极,结合无线传输技术,实现对清醒动物神经化学的长期监测,无疑将推动脑神经生理和病理的深入研究和探索; 再次,随着科学技术的不断进步,脑科学的发展正在迈入新的纪元,其与各领域的界线逐渐模糊。最后,有效利用新型生物电化学传感器探索脑神经活动的化学基础将是人类研究脑、认识脑的关键。总之,虽然利用电化学生物传感器在活体层次精准获取化学信号研究充满了挑战,但相信生物电化学传感器在脑神经分析化学研究领域仍具有良好的发展前景。
References
1 Alivisatos A P, Chun M, Church G M, Deisseroth K, Donoghue J P, Greenspan R J, McEuen P L, Roukes M L, Sejnowski T J, Weiss P S, Yuste R. Science, 2013, 339(6125): 1284-1285
2 Stuart J N, Hummon A B, Sweedler J V. Anal. Chem., 2004, 76(7): 121A-128A
3 Andersson S, Klinteberg C, Svanberg K, Svanberg S. Phys. Med. Biol., 1997, 42(5): 815-824
4 McDonnell L A, Heeren R M. Mass Spectrom. Rev., 2007, 26(4): 606-643
5 Choi I Y, Lee S P, Guilfoyle D N, Helpern J A. Neurochem. Res., 2003, 28(7): 987-1001
6 Fox M D, Raichle M E. Nat. Rev. Neurosci., 2007, 8(9): 700-711
7 Greco J B, Sakaie K E, Aminipour S, Lee P L, Chang L L, He J, Westmoreland S, Lackner A A, Gonzalez R G. J. Med. Primatol., 2002, 31(4 5): 228-236
8 Gambhir S S. Nat. Rev. Cancer, 2002, 2(9): 683-693
9 Wagner A, Mahrholdt H, Holly T A, Elliott M D, Regenfus M, Parker M, Klocke F J, Bonow R O, Kim R J, Judd R M. Lancet, 2003, 361(9355): 374-379
10 Chai X, Zhou X, Zhu A, Zhang L, Qin Y, Shi G, Tian Y. Angew. Chem. Int. Ed., 2013, 52(31): 8129-8133
11 Luo Y, Zhang L, Liu W, Yu Y, Tian Y. Angew. Chem. Int. Ed., 2015, 54(47): 14053-14056
12 Yu P, He X, Mao L. Chem. Soc. Rev., 2015, 44(17): 5959-5968
13 Wang Y, Mao L. Electroanalysis, 2016, 28(2): 265-276
14 Liu X, Xiao T, Wu F, Shen M Y, Zhang M, Yu H H, Mao L. Angew. Chem. Int. Ed., 2017, 56(39): 11802-11806
15 Wu F, Yu P, Mao L. Curr. Opin. Electrochem., 2017, 5(1): 152-157
16 Wu F, Yu P, Mao L. Chem. Soc. Rev., 2017, 46(10): 2692-2704
17 Cheng H, Li L, Zhang M, Jiang Y, Yu P, Ma F, Mao L. TrAC Trends Anal. Chem., 2018, 109: 247-259
18 Wu F, Yu P, Mao L. ACS Omega, 2018, 3(10): 13267-13274
19 Zhang L, Tian Y. Acc. Chem. Res., 2018, 51(3): 688-696
20 Xiao T, Wang Y, Wei H, Yu P, Jiang Y, Mao L. Angew. Chem. Int. Ed., 2019, 58(20): 6616-6619
21 Cheer J F, Heien M L, Garris P A, Carelli R M, Wightman R M. Proc. Natl. Acad. Sci. USA, 2005, 102(52): 19150-19155
22 Xiang L, Yu P, Hao J, Zhang M, Zhu L, Dai L, Mao L. Anal. Chem., 2014, 86(8): 3909-3914
23 Yue X, Zhu Z, Zhang M, Ye Z. Anal. Chem., 2015, 87(3): 1839-1845
24 Ren L, Pour M D, Majdi S, Li X, Malmberg P, Ewing A G. Angew. Chem. Int. Ed., 2017, 56(18): 4970-4975
25 Wang K, Zhao X, Li B, Wang K, Zhang X, Mao L, Ewing A, Lin Y. Anal. Chem., 2017, 89(17): 8683-8688
26 Wang S, Liu X, Zhang M. Anal. Chem., 2017, 89(10): 5382-5388
27 Li X, Dunevall J, Ewing A G. Faraday Discuss., 2018, 210(0): 353-364
28 Shin M, Venton B J. Anal. Chem., 2018, 90(17): 10318-10325
29 Smith S K, Gosrani S P, Lee C A, McCarty G S, Sombers L A. Anal. Chem., 2018, 90(21): 12994-12999
30 Wilson L R, Panda S, Schmidt A C, Sombers L A. Anal. Chem., 2018, 90(1): 888-895
31 Yang C, Cao Q, Puthongkham P, Lee S T, Ganesana M, Lavrik N V, Venton B J. Angew. Chem. Int. Ed., 2018, 57(43): 14255-14259
32 Robinson D L, Hermans A, Seipel A T, Wightman R M. Chem. Rev., 2008, 108(7): 2554-2584
33 Wilson G S, Johnson M A. Chem. Rev., 2008, 108(7): 2462-2481
34 Clark L C, Lyons C. Ann. NY Acad. Sci., 1962, 102:29-45
35 Wilson G S, Ammam M. FEBS J., 2007, 274(21): 5452-5461
36 Wilson G S, Hu Y. Chem. Rev., 2000, 100(7): 2693-2704
37 Xiao T, Wu F, Hao J, Zhang M, Yu P, Mao L. Anal. Chem., 2017, 89(1): 300-313
38 Nichols S P, Koh A, Storm W L, Shin J H, Schoenfisch M H. Chem. Rev., 2013, 113(4): 2528-2549
39 Wang J. Chem. Rev., 2008, 108(2): 814-825
40 Burmeister J J, Moxon K, Gerhardt G A. Anal. Chem., 2000, 72(1): 187-192
41 Moon B U, de Vries M G, Cordeiro C A, Westerink B H, Verpoorte E. Anal. Chem., 2013, 85(22): 10949-10955
42 Anzai J, Takeshita H, Kobayashi Y, Osa T, Hoshi T. Anal. Chem., 1998, 70(4): 811-817
43 Baker K L, Bolger F B, Lowry J P. Analyst, 2015, 140(11): 3738-3745
44 Li C, Limnuson K, Wu Z, Amin A, Narayan A, Golanov E V, Ahn C H, Hartings J A, Narayan R K. Biosens. Bioelectron., 2016, 77: 62-68
45 Chatard C, Sabac A, Moreno Velasquez L, Meiller A, Marinesco S. ACS Cent. Sci., 2018, 4(12): 1751-1760
46 Rutherford E C, Pomerleau F, Huettl P, Stromberg I, Gerhardt G A. J. Neurochem., 2007, 102(3): 712-722
47 Burmeister J J, Gerhardt G A. Anal. Chem., 2001, 73(5): 1037-1042
48 Burmeister J J, Pomerleau F, Palmer M, Day B K, Huettl P, Gerhardt G A. J. Neurosci. Methods, 2002, 119(2): 163-171
49 Delgado J M R, Defeudis F V, Roth R H, Ryugo D K, Mitruka B M. Arch. Int. Pharmacodyn. Ther., 1972, 198(1): 9-21
50 LYU Yang, ZHANG Ya Wen, TAN Lei, JI Wen Liang, YU Ping, MAO Lan Qun, ZHOU Fang. Chinese J. Anal. Chem., 2017, 45(11): 1595-1599
呂 扬, 张雅文, 谭 磊, 纪文亮, 于 萍, 毛兰群, 周 方. 分析化学, 2017, 45(11): 1595-1599
51 Zhang M, Yu P, Mao L. Acc. Chem. Res., 2012, 45(4): 533-543
52 Wilson G S, Gifford R. Biosens. Bioelectron., 2005, 20(12): 2388-2403
53 Rogers M L, Feuerstein D, Leong C L, Takagaki M, Niu X, Graf R, Boutelle M G. ACS Chem. Neurosci., 2013, 4(5): 799-807
54 Varner E L, Leong C L, Jaquins Gerstl A, Nesbitt K M, Boutelle M G, Michael A C. ACS Chem. Neurosci., 2017, 8(8): 1779-1788
55 Niwa O, Torimitsu K, Morita M, Osborne P, Yamamoto K. Anal. Chem., 1996, 68(11): 1865-1870
56 Osborne P G, Niwa O, Yamamoto K. Anal. Chem., 1998, 70(9): 1701-1706
57 Mao L, Yamamoto K. Anal. Chim. Acta, 2000, 415(1 2): 143-150
58 Mao L, Yamamoto K. Talanta, 2000, 51(1): 187-195
59 Mao L, Osborne P G, Yamamoto K, Kato T. Anal. Chem., 2002, 74(15): 3684-3689
60 Lin Y, Liu K, Yu P, Xiang L, Li X, Mao L. Anal. Chem., 2007, 79(24): 9577-9583
61 Cass A E, Davis G, Francis G D, Hill H A, Aston W J, Higgins I J, Plotkin E V, Scott L D, Turner A P. Anal. Chem., 1984, 56(4): 667-671
62 Xiang L, Zhang Z, Yu P, Zhang J, Su L, Ohsaka T, Mao L. Anal. Chem., 2008, 80(17): 6587-6593
63 Hu J, Turner A P. Anal Lett., 1991, 24(1): 15-24
64 Forrow N J, Walters S J. Biosens. Bioelectron., 2004, 19(7): 763-770
65 Bartlett P N, Booth S, Caruana D J, Kilburn J D, Santamaria C. Anal. Chem., 1997, 69(4): 734-742
66 Zhang Z, Hao J, Xiao T, Yu P, Mao L. Analyst, 2015, 140(15): 5039-5047
67 Suzuki A, Mano N, Tsujimura S. Electrochim. Acta, 2017, 232: 581-585
68 Zhuang X, Wang D, Lin Y, Yang L, Yu P, Jiang W, Mao L. Anal. Chem., 2012, 84(4): 1900-1906
69 Wang X, Li Q, Xu J, Wu S, Xiao T, Hao J, Yu P, Mao L. Anal. Chem., 2016, 88(11): 5885-5891
70 Lin Y, Zhu N, Yu P, Su L, Mao L. Anal. Chem., 2009, 81(6): 2067-2074
71 Lin Y, Yu P, Hao J, Wang Y, Ohsaka T, Mao L. Anal. Chem., 2014, 86(8): 3895-3901
72 Yu P, Zhou H, Cheng H, Qian Q, Mao L. Anal. Chem., 2011, 83(14): 5715-5720
73 Ma W, Jiang Q, Yu P, Yang L, Mao L. Anal. Chem., 2013, 85(15): 7550-7557
74 WANG Ting Ting, ZHANG Jie, WANG Shen Shuai, WANG Xiu Yun. Chinese J. Anal. Chem., 2019, 47(7): 1021-1028
王婷婷, 張 杰, 王沈帅, 王秀云. 分析化学, 2019, 47(7): 1021-1028
75 Huang P, Mao J, Yang L, Yu P, Mao L. Chem. Eur. J., 2011, 17(41): 11390-11393
76 Lu X, Cheng H, Huang P, Yang L, Yu P, Mao L. Anal. Chem., 2013, 85(8): 4007-4013
77 Solomon E I, Sundaram U M, Machonkin T E. Chem. Rev., 1996, 96(7): 2563-2606
78 Wise R A. Nat. Rev. Neurosci., 2004, 5(6): 483-494
79 Xiang L, Lin Y, Yu P, Su L, Mao L. Electrochim. Acta, 2007, 52(12): 4144-4152
80 Lin Y, Zhang Z, Zhao L, Wang X, Yu P, Su L, Mao L. Biosens. Bioelectron., 2010, 25(6): 1350-1355
81 Le Goff A, Holzinger M, Cosnier S. Cell. Mol. Life Sci., 2015, 72(5): 941-952
82 Zheng W, Li Q, Su L, Yan Y, Zhang J, Mao L. Electroanalysis, 2006, 18(6): 587-594
83 Wu F, Su L, Yu P, Mao L. J. Am. Chem. Soc., 2017, 139(4): 1565-1574
84 Han Z, Zhao L, Yu P, Chen J, Wu F, Mao L. Electrochem. Commun., 2019, 101: 82-87
85 Wu F, Cheng H, Wei H, Xiong T, Yu P, Mao L. Anal. Chem., 2018, 90(21): 13021-13029
86 Wu F, Yu P, Yang X, Han Z, Wang M, Mao L. J. Am. Chem. Soc., 2018, 140(40): 12700-12704
87 Song S, Wang L, Li J, Zhao J, Fan C. TrAC Trends Anal. Chem., 2008, 27(2): 108-117
88 Nakatsuka N, Yang K A, Abendroth J M, Cheung K M, Xu X, Yang H, Zhao C, Zhu B, Rim Y S, Yang Y, Weiss P S, Stojanovic M N, Andrews A M. Science, 2018, 362(6412): 319-324
89 Yu P, He X, Zhang L, Mao L. Anal. Chem., 2015, 87(2): 1373-1380
90 Keighron J D, Wigstrom J, Kurczy M E, Bergman J, Wang Y, Cans A S. ACS Chem. Neurosci., 2015, 6(1): 181-188
91 Patel B A, Rogers M, Wieder T, O'Hare D, Boutelle M G. Biosens. Bioelectron., 2011, 26(6): 2890-2896
92 Varju P, Katarova Z, Madarasz E, Szabo G. Cell Tissue Res., 2001, 305(2): 239-246
93 Niwa O, Horiuchi T, Kurita R, Torimitsu K. Anal. Chem., 1998, 70(6): 1126-1132
94 Burmeister J J, Pomerleau F, Huettl P, Gash C R, Werner C E, Bruno J P, Gerhardt G A. Biosens. Bioelectron., 2008, 23(9): 1382-1389
95 ZHAO Fan, SHI Guo Yue, TIAN Yang. Chinese J. Anal. Chem., 2019, 47(3): 347-354
趙 凡, 施国跃, 田 阳. 分析化学, 2019, 47(3): 347-354
96 Zhang Z, Xiao T, Hao J, Yu P, Ohsaka T, Mao L. Electroanalysis, 2015, 27(10): 2406-2411
97 Llaudet E, Botting N P, Crayston J A, Dale N. Biosens. Bioelectron., 2003, 18(1): 43-52
98 Llaudet E, Hatz S, Droniou M, Dale N. Anal. Chem., 2005, 77(10): 3267-3273
99 Gourine A V, Llaudet E, Dale N, Spyer K M. Nature, 2005, 436(7047): 108-111
100 Zhang K, He X, Liu Y, Yu P, Fei J, Mao L. Anal. Chem., 2017, 89(12): 6794-6799
101 Tybrandt K, Kollipara S B, Berggren M. Sens. Actuators B, 2014, 195: 651-656
102 Guo S, Yan H, Wu F, Zhao L, Yu P, Liu H, Li Y, Mao L. Anal. Chem., 2017, 89(23): 13008-13015
103 Ma W, Mao J, Yang X, Pan C, Chen W, Wang M, Yu P, Mao L, Li Y. Chem. Commun., 2018, 55(2): 159-162
104 Liu M, Johnston M B, Snaith H J. Nature, 2013, 501(7467): 395-398
Advances in Electrochemical Biosensors for in Vivo Analysis
WEI Huan1,2, WU Fei1,2, YU Ping1,2, MAO Lan Qun*1,2
1(Beijing National Laboratory for Molecular Science, Key Laboratory of Analytical Chemistry for Living Biosystems,
Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, China)
2(University of Chinese Academy of Sciences, Beijing 100049, China)
Abstract As the chemical species build the essential basis for mediating and modulating neurotransmission that determines various physiological and pathological states of the central nervous system (CNS), the fundamental research on neurochemistry, especially the investigation of the correlation between neurochemical dynamics and activities of vesicles, single cells, neural circuits and the entire brain, has attracted increasing attention due to its significance in understanding the brain function. Electrochemical analytical methods have made tremendous achievements for in vivo and online continuous monitoring of neurotransmitters and neuromodulators. Among them, the methods utilizing enzymes or aptamers as the biorecognition elements and rationally designing electrode surfaces/interfaces to construct the electrochemical biosensors with high selectivity and high sensitivity undoubtedly provide attractive approaches to quantitative monitoring of brain chemistry. This review mainly focuses on the recent advances in electrochemical biosensors for in vivo analysis.
Keywords Electrochemical biosensors; In vivo analysis; Online electrochemical analysis; Brain chemistry; Review
