自由装填推进剂用含醛基/烯丙基芳氧基聚磷腈包覆材料研究(Ⅰ):制备、硫化特性及力学性能
2019-11-11曹继平赵凤起杨士山
曹继平,肖 啸,魏 乐,赵凤起,2,杨士山
(1.西安近代化学研究所,陕西 西安 710065; 2.西安近代化学研究所燃烧与爆炸技术重点实验室,陕西 西安 710065)
引 言
包覆层是自由装填式推进剂装药的重要组成部分,主要起限制推进剂药柱燃烧面积、确保发动机按照设计的内弹道参数工作的作用[1-2]。包覆层通常由高分子材料、无机或有机功能填料及添加剂组成,具有与固体推进剂相容性优异、力学性能匹配、烧蚀性能优良等特点[3]。目前,适用于自由装填式装药的包覆材料主要包括以醋酸纤维素、乙基纤维素、聚乙烯、聚苯乙烯等为代表的热塑性包覆材料,以不饱和聚酯树脂、聚氨酯、环氧树脂、硅橡胶等为代表的热固性树脂包覆材料和以三元乙丙橡胶为代表的橡胶类材料。然而,随着固体推进剂不断向高能化方向发展,自由装填式推进剂装药对包覆层的综合性能提出了日趋严苛的技术要求。
芳氧基取代聚磷腈橡胶因其独特的分子结构和发烟量低、耐烧蚀等级高、抗增塑剂迁移等诸多优点,是未来高燃速低特征信号自由装填式推进剂装药理想的包覆层材料[4]。自20世纪70年代以来,国外针对芳氧基聚磷腈的合成及其在固体火箭发动机绝热层方面的应用进行了研究,并开发出了多种芳氧基聚磷腈绝热层配方。研究结果表明,芳氧基聚磷腈为基体的绝热层配方具有如下优势:(1)低发烟性。根据美国专利[5]介绍,芳氧基聚磷腈绝热层配方的烟雾试验达到了北约国家烟雾测试标准的A级;(2)耐烧蚀性。法国Thiokol公司的研究结果表明[6],芳氧基聚磷腈绝热层配方具有优异的耐烧蚀性,适合作为高燃速推进剂发动机绝热层材料使用;(3)阻隔性。美国Ethyl公司公布了一种高交联密度的含有不饱和双键的芳氧基聚磷腈包覆层配方[7-8],该配方具有优异的抗增塑剂迁移性能,其对推进剂中常用小组分的吸收全部低于HTPB材料,对硝酸酯增塑剂的吸收在六周之后仍然小于2%;(4)自熄性。芳氧基聚磷腈绝热层配方在明火中点燃并使其燃烧15s,取出后3s自熄。我国王志锋等[9-11]针对芳氧基聚磷腈弹性体的合成及其在自由装填式推进剂装药包覆领域的应用进行了较为系统的研究,突破了六氯环三磷腈开环聚合、弹性体纯化、包覆层配方设计与成型工艺以及与改性双基推进剂界面粘接等关键技术,为工程化应用奠定了基础[9-11]。研究结果发现,以苯酚和对乙基苯酚取代的芳氧基聚磷腈弹性体交联机理呈现大分子链耦合形成交联网络的饱和橡胶特征,因此硫化橡胶力学性能不高。
鉴于芳氧基聚磷腈的性能优势以及在自由装填推进剂装药包覆层领域的应用潜力,本研究通过向聚磷腈分子主链上引入具有交联反应活性的醛基苯氧基和烯丙基苯氧基,合成新型的醛基/烯丙基混合取代交联型芳氧基聚磷腈(PDPP),并以此为基体开展了自由装填推进剂装药包覆层配方设计、硫化特性及力学性能研究,为其实际应用提供参考。
1 试 验
1.1 材 料
六氯环三磷腈,分析纯,山东淄博蓝印化工有限公司;4-羟基苯甲醛,分析纯,山东鲁科化工有限公司,使用前用水重结晶后真空干燥;2-烯丙基苯酚,分析纯,百灵威科技有限公司;金属钠、苯酚、四氢呋喃、二水合硫酸钙,分析纯,成都市科龙化工试剂厂;α-氯代萘,分析纯,阿拉丁试剂公司;氨基磺酸,分析纯,上海谱振生物科技有限公司;白炭黑、氢氧化铝、硫化剂,工业品,天津化学试剂厂;硬脂酸,分析纯,西安化工试剂厂;磷酸三氯乙酯,分析纯,上海彭浦化工试剂厂。
1.2 仪器及测试方法
红外光谱采用美国Nicolet傅里叶变换红外光谱仪测定;密炼过程采用Haake Polylab流变仪密炼机头;凝胶渗透色谱采用美国PL GPC-50型凝胶渗透色谱仪测定;橡胶硫化采用西北橡胶厂机械分厂的WDl600平板硫化机;硫化特性参数采用MDR 2000E型橡胶硫化仪测试(执行GB/T16584-1996);静态力学性能采用美国Instron公司Instron 4505型万能材料试验机测试(执行GB/T528-2009);动态机械热分析(DMTA)用Rheometric DMTA-A型动态热分析仪测试;线膨胀系数采用PZY-Ⅲ-14/17型线膨胀系数测试仪测试(执行GJB770B-2005 401.2)。
1.3 PDPP的合成
以六氯环三磷腈为原料,二水合硫酸钙和氨基磺酸为催化剂,在α-氯代萘体系中经开环聚合反应得到中间体聚二氯磷腈;将4-羟基苯甲醛和2-烯丙基苯酚按一定质量比混合,与金属钠反应得到相应的酚钠盐混合物;聚二氯磷腈再与苯酚钠及所得的两种酚钠盐混合物进行亲核取代反应,得到4种PPDP。合成路线如下:
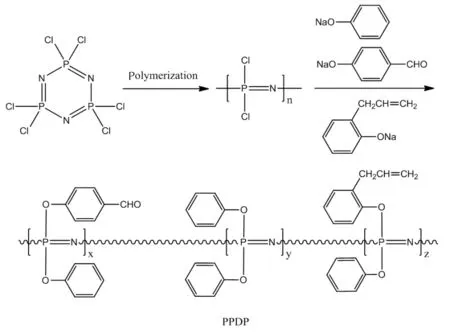
1.3.1 聚二氯磷腈的合成
称取24g(69mmol)六氯环三磷腈、50.8mg(0.52mmol)氨基磺酸、45mg(0.26mmol)二水合硫酸钙和25mLα-氯代萘加入250mL配置油浴、回流装置和氮气保护装置的三口反应瓶中。搅拌下缓慢升温至220℃,并恒温反应至体系黏度发生明显变化时立即终止反应。将所得反应液缓慢倒入盛有400mL正庚烷的烧杯中,不断搅拌至液相呈现透明,然后移出液相混合物,将所得白色黏性沉淀物用150mL无水四氢呋喃溶解(不溶物过滤除去),再将所得四氢呋喃溶液缓慢倒入盛有400mL正庚烷的烧杯中,不断搅拌至液相呈现透明并沉淀产生灰白色黏性物。重复上述步骤3次后,将所得灰白色黏性物在室温条件下真空干燥6 h,得到灰白色透明橡胶体,收率35%~45%。
IR(KBr),ν(cm-1):3432,2923,1635,1567,1504,1230,969,791,765,665,575。
由于聚二氯磷腈分子结构中含有大量的活性氯原子,在进行凝胶渗透色谱测定分子质量以及分子质量分布时容易发生色谱柱吸附堵塞,故对其分子质量及分子质量分布不进行测试。
1.3.2 PDPP的合成
按表1的原料配比制备4种PDPP样品。称取4-羟基苯甲醛、2-烯丙基苯酚和350mL四氢呋喃加入到500mL配置机械搅拌、温度计、冷凝装置和冰盐浴的三口反应瓶中,搅拌至固体完全溶解后将反应体系冷却至0~5℃,缓慢分批加入定量的金属钠丝。加毕,室温下搅拌反应6h,再升温至65℃并恒温反应24h。反应完成后,过滤,收集滤液备用。再按相同的操作步骤制备苯酚钠,备用。
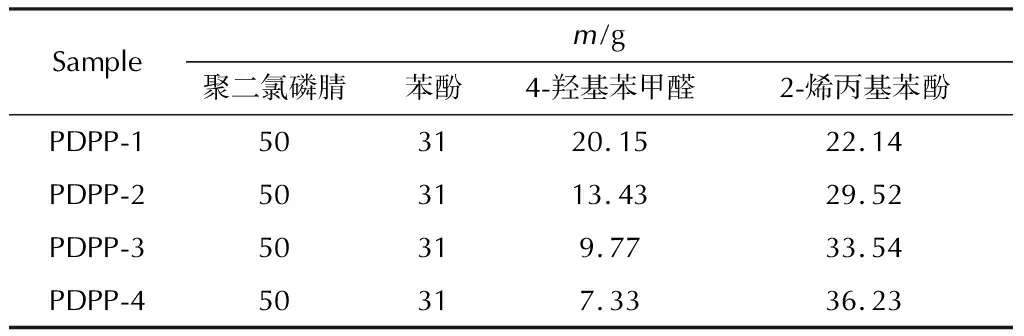
表1 制备PDPP的原料配比
将一定量聚二氯磷腈溶于无水四氢呋喃中配置成溶液(溶液质量分数以聚二氯磷腈完全溶解为准),然后向其中缓慢加入定量的苯酚钠,加完后30℃反应3h;然后向反应体系中滴加所得的4-羟基苯甲醛钠盐和2-烯丙基苯酚钠盐的四氢呋喃混合溶液,滴加过程中控制反应体系温度不高于30℃。滴毕,室温下反应8h,然后50℃下恒温反应12h,再升温至65℃恒温反应24h。反应完成后,减压回收约1/3的溶剂,将剩余反应混合液缓慢倒入大量的蒸馏水中,不断搅拌至灰白色橡胶体完全析出。将所得橡胶体用80℃热水浸泡并不断换水,直至水相呈无色透明。将所得橡胶体真空80℃干燥至恒重,即得目标产物。
表2为4种PDPP的红外光谱和凝胶渗透色谱分析结果。
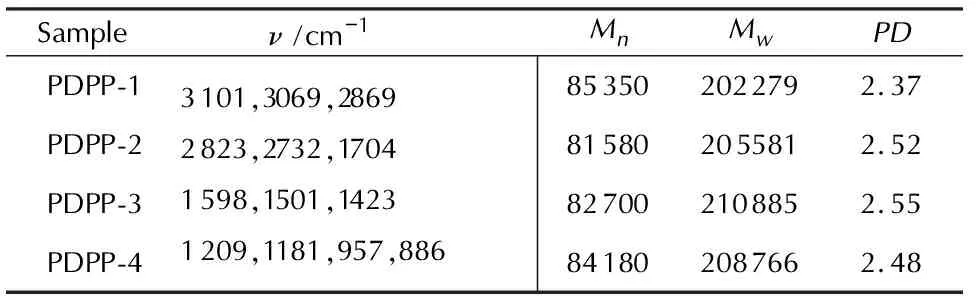
表2 PDPP的红外光谱和凝胶渗透色谱分析结果
1.4 包覆层制备
包覆层基础配方为:氧化锌5g,硬脂酸0.8g,过氧化物1g,硫0.8g,炭黑1g,磷酸三氯乙酯2.5g,氢氧化铝10g,芳纶1414短纤维(6.0±0.2mm)5g,碳酸钙1g。在基础配方中分别添加100g PDPP1~PDPP4制成4种包覆层样品,分别记为样品1~样品4。
包覆层硫化工艺参数为:150℃、压强8MPa 下硫化20min。
2 结果与讨论
2.1 PDPP合成思路
本研究旨在合成一种含有醛基和烯丙基的芳氧基聚磷腈弹性体,利用醛基之间的安息香缩合反应和烯丙基(即不饱和双键)的自由基聚合反应提高材料的交联密度,从而提高包覆层材料的力学强度和耐热、耐烧蚀性能。据文献[12-16]中的研究结论,聚磷腈的物化性质取决于所选取代基的类型:苯氧基作为单一取代基,所得的苯氧基聚磷腈为橡胶弹性体;引入部分烯丙基芳氧基后,所得混合取代聚合物的玻璃化转变温度升高,但物理聚集态仍为弹性体;而有关在聚磷腈主链结构上引入醛基芳氧基尚未见文献报道。因此,本研究中PDPP的合成思路为:利用聚二氯磷腈取代反应过程中可快速形成单取代结构的反应特性,通过调整投料顺序和投料比,首先形成苯氧基单取代的聚磷腈中间体,然后再向分子主链上引入醛基苯氧基和烯丙基苯氧基取代侧基结构,从而合成出含有醛基和烯丙基的混合取代芳氧基聚磷腈弹性体。
2.2 包覆层的硫化特性分析
包覆层样品1~样品4的硫化特性曲线如图1所示,硫化特性参数见表3。
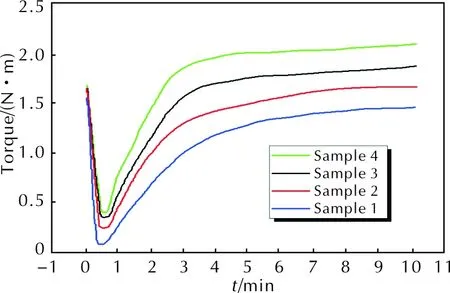
图1 包覆层样品1~样品4的硫化特性曲线Fig. 1 Vulcanization characteristic curves of sample 1—4
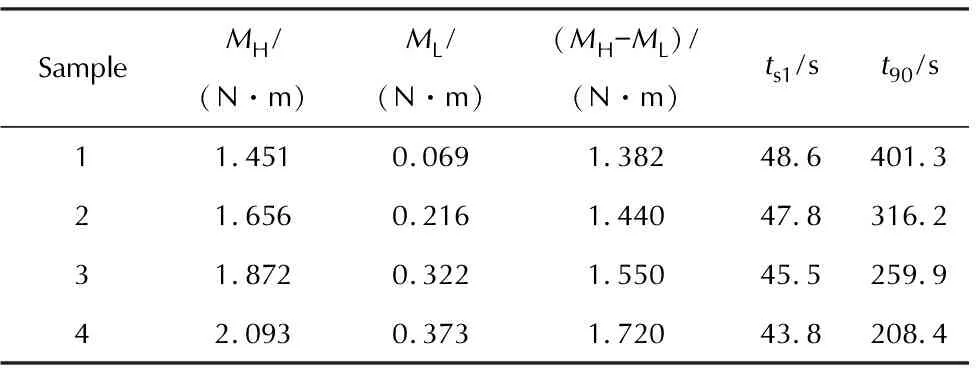
SampleMH/(N·m)ML/(N·m)(MH-ML)/(N·m)ts1/st90/s11.4510.0691.38248.6401.321.6560.2161.44047.8316.231.8720.3221.55045.5259.942.0930.3731.72043.8208.4
由图1和表3可知,随着配方中醛基与烯丙基质量比的降低:(1)焦烧时间(ts1)逐渐减小,说明醛基和烯丙基的配比与胶料的操作安全性有关,醛基含量越低(即烯丙基含量相对越高),自由基交联网络的生成速率越快,安全操作时间则越短;(2)正硫化时间(t90)逐渐缩短,说明烯丙基浓度越高,自由基聚合速率越快,硫化速率则越高;(3)最低转矩(ML)、最高转矩(MH)以及转矩差(MH-ML)不断升高,说明胶料的流动性随聚磷腈主链上烯丙基芳氧基取代度的提高而逐渐变差,材料的交联密度增大,硬度提高。此变化规律与文献[5-6]中有关聚磷腈物化性质和取代基关系的结论基本一致。
2.3 包覆层样品的力学性能分析
2.3.1 应力—应变及线膨胀系数分析
图2分别为样品在高温(+50℃)、低温(-40℃)和常温(+20℃)条件下的应力―应变曲线,表4为包覆层样品在不同温度条件下的力学性能数据。

图2 不同温度下样品1~样品4的应力―应变曲线Fig. 2 Stress—strain curvs of sample 1-4 at different temperatures
由图2可见,在+50℃、+20℃和-40℃条件下4种样品应力—应变曲线斜率不断增加,直至测试样条断裂,整个拉伸过程中并未发生屈服,测试样品也未出现局部径缩现象,且拉伸后的材料在撤掉外力后能很快恢复拉伸前的形状,说明在测试温度下4种样品在断裂之前均处于弹性阶段。而在-40℃条件下时,4种样品均处于玻璃态,拉伸过程中会发生明显的脆断现象。
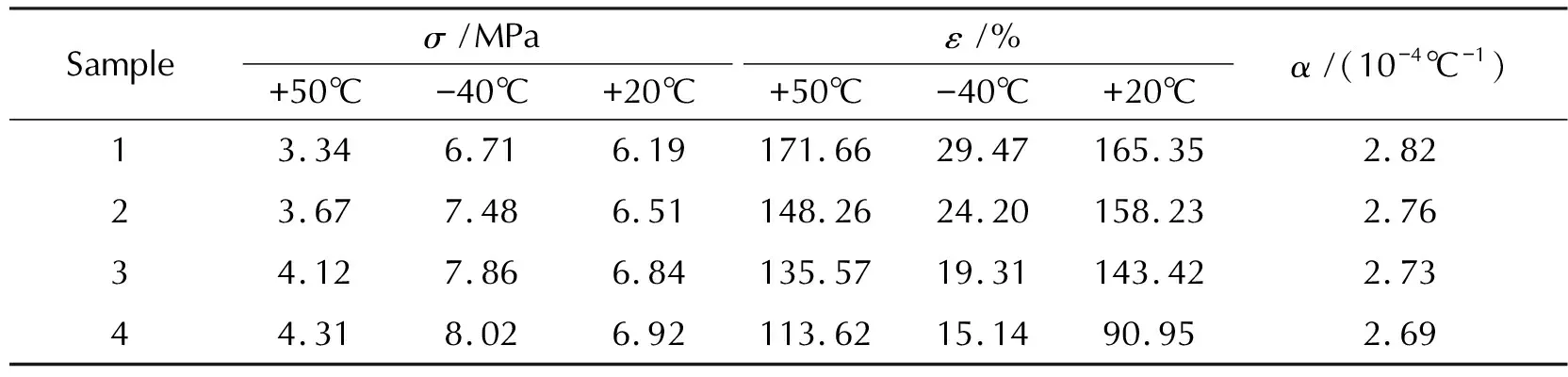
表4 样品1~样品4在不同温度下的拉伸强度、延伸率及线膨胀系数Table 4 Tensile strengths, elongations and linear expansion coefficients of sample 1—4 at different temperatures
由表4中数据可知,相同的测试温度条件下,随着配方中醛基与烯丙基质量比的降低,拉伸强度不断提高,断裂延伸率和线膨胀系数不断降低。这是因为拉伸强度、延伸率和线膨胀系数与材料的平均键强度相关,平均键强度越高,拉伸强度越高,延伸率和线膨胀系数越低。因此,随着烯丙基含量的不断增加,硫化胶交联网络结构中所含的由自由基聚合产生的碳碳单键交联点含量相对提高,由醛基缩合产生的醚键交联点含量相对降低,而碳碳单键的键强度高于醚键,导致拉伸强度升高,延伸率和线膨胀系数降低。此外,双基推进剂的线膨胀系数约为0.8×10-4~1.0×10-4℃-1,本研究的包覆层试样的线膨胀系数为2.69×10-4~2.82×10-4℃-1,明显高于双基推进剂的线膨胀系数,说明包覆装药在环境温度变化时包覆层能够随着推进剂药柱的热胀冷缩而自适应变形,确保装药包覆可靠。
2.3.2 动态机械热分析
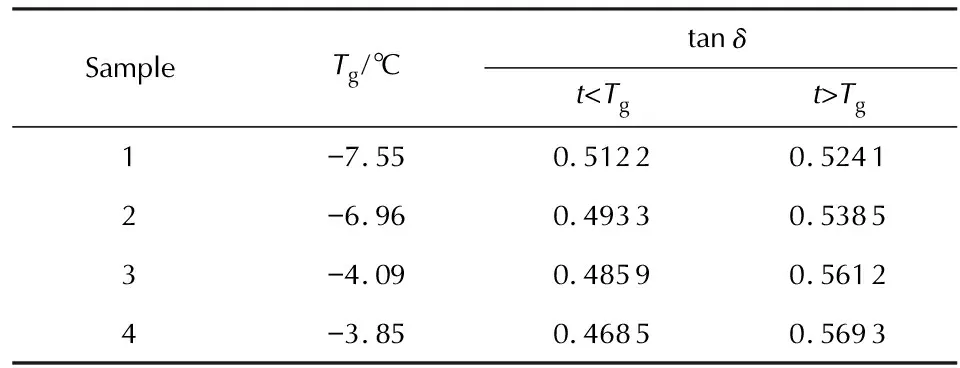
利用动态机械热分析(DMTA)测定了PDPP和包覆层样品在交变应力(或应变)作用下应变(或应力)响应随温度或频率的变化关系,包括玻璃化转变温度(Tg)、贮能模量(E′)、损耗模量(E″)和损耗因子(tanδ=E″/E′)。图3和图4分别为4种PDPP样品和4种包覆层样品的DMTA曲线,损耗因子平均值见表5。
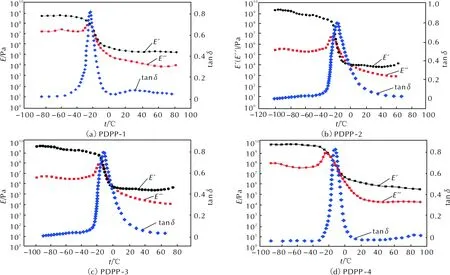
图3 PDPP-1~PDPP-4的DMTA曲线Fig.3 DMTA curves of PDPP-1—PDPP-4
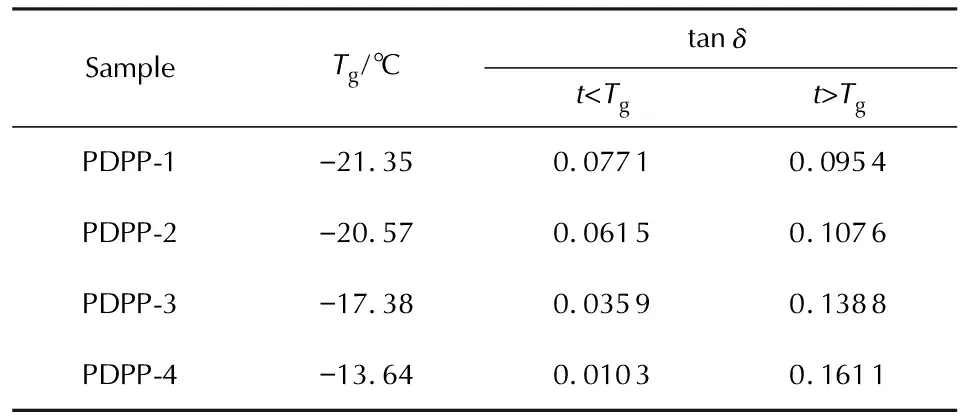
表5 PDPP-1~PDPP-4的Tg和tanδ均值
由图3和表5中数据可知,随着生胶中烯丙基含量的不断提高,tanδ—t曲线逐渐向高温方向移动,Tg逐渐升高。这种影响在微观上归因于烯丙基苯氧基之间、烯丙基苯氧基与苯氧基之间以及烯丙基与聚磷腈主链之间的空间位阻效应对分子链段运动以及分子链构象转变的影响[7]。结合文献中Allcock等[15]建立的聚磷腈分子模型以及分子链运动规律可知,未取代的聚二氯磷腈分子链单元不同构象间的位垒小(小于4.187kJ/mol),分子链不需要获得较大能量就可以自由旋转,则表现出良好的柔顺性。而芳氧基取代的聚磷腈分子链单元不同构象间的位垒较高(21.814kJ/mol),分子链结构单元难以克服较高的位垒,故苯氧基取代的聚磷腈分子链表现出较强的刚性,玻璃化温度相应提高。因此,根据以上理论可以推断,由于2-烯丙基苯氧基的空间位阻较4-醛基苯氧基的空间位阻更大,随着烯丙基苯氧基取代度的增加,分子链结构单元运动需克服的位垒更高,分子链刚性随之增强,在宏观上则表现为玻璃化转变温度提高。
此外,从图3和表5还可以看出,在t 当t>Tg时,tanδ则随烯丙基苯氧基取代度的提高而增加。这是因为随着温度进一步升高,分子间作用力逐渐减弱,被冻结的高分子链段开始逐渐运动,高分子链可以拉直或卷曲直至出现构象改变,材料达到柔软且富有弹性的高弹态,贮能模量(E′)迅速发生数量级的骤变。此时,随着烯丙基苯氧基取代度的提高,侧基空间位阻增大,分子链的整体位移会引起内摩擦阻力的增大,tanδ呈现出增大的变化趋势。 图4 包覆层样品1~样品4的DMTA曲线Fig. 4 DMTA curves of sample 1—4 表6 包覆层样品1~样品4的Tg和tanδ均值 由图4和表6中数据可知,随着配方中烯丙基含量的不断提高,tanδ—t曲线逐渐向高温方向移动,Tg逐渐升高。这种变化趋势与未经硫化的生胶基本一致。值得注意的是,生胶在硫化成型前须经密炼、填充处理,密炼过程中的剪切作用使聚合物因部分水解反应产生的P—O—P键交联体系被破坏而发生解交联反应,导致生胶交联密度下降,交联对链段运动的束缚作用逐步减弱,在一定程度上强化了分子链的自由运动能力,在热学性质上表现为玻璃化转变温度降低。然而,硫化成型过程中,胶料内部因烯丙基的自由基交联反应以及醛基的缩合反应重新构建了更加致密的交联网络,硫化胶的玻璃化转变温度较生胶有明显提高,而密炼-解交联作用所产生的玻璃化转变温度降低作用则影响甚微。因此,影响硫化胶玻璃化转变温度的因素微观上仍可归咎于烯丙基苯氧基之间的空间位阻效应以及烯丙基与聚磷腈主链之间的空间位阻效应对分子链段运动以及分子链构象转变的影响,故包覆层样品的玻璃化温度变化趋势与PDPP体系基本一致。此外,对比包覆层样品和PDPP的损耗因子tanδ可以发现,包覆层样品的tanδ明显高于生胶。这是因为tanδ值表示损耗能量的大小,tanδ均值越大,表明链段松弛运动导致的大分子层内摩擦越大,相对损耗能力越大。硫化胶因自身交联密度较大,分子链自由运动时受到的摩擦力较大而导致tanδ均值较大。 (1)以六氯环三磷腈、苯酚、4-羟基苯甲醛和2-烯丙基苯酚为原料合成出4种含醛基/烯丙基交联型芳氧基聚磷腈。 (2)随着包覆层体系中醛基/烯丙基摩尔比的减小,胶料的焦烧时间(ts1)和正硫化时间(t90)缩短,最低转矩(ML)、最高转矩(MH)以及转矩差(MH-ML)逐渐升高。 (3)在高温(+50℃)、低温(-40℃)和常温(+20℃)静态拉伸条件下,样品1~4在断裂之前均处于弹性阶段,其应力―应变曲线斜率不断增加,整个拉伸过程中并未发生屈服,测试样品也未出现局部径缩现象。 (4)随着生胶烯丙基含量的不断提高,tanδ-t曲线逐渐向高温方向移动,Tg逐渐升高。与PDPP相比,包覆层样品的玻璃化转变温度和tanδ有所提高,且玻璃化转变温度与tanδ的变化趋势与PDPP体系基本一致。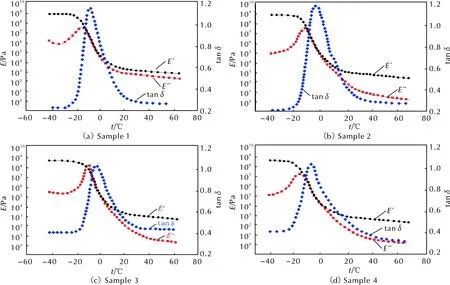
3 结 论
