Comparison of COSMO-SAC Model and DFT Quantum Chemical Model in Molecular Design of IL Extractant
2019-10-31FangJingWangYijingZhangShutingZhengWenwenLiHaoLiChunli
Fang Jing; Wang Yijing; Zhang Shuting; Zheng Wenwen; Li Hao; Li Chunli
(School of Chemical Engineering, Hebei University of Technology, Tianjin 300130)
Abstract: ΙLs agents with better extraction efficiency for desulfurization were screened by simulation in this paper. The COSMO-SAC model and the density functional theory (DFT) were used to select the prospective solvents from a wide variety of ionic liquids suitable for extractive distillation. The COSMO-SAC model and the density functional theory (DFT)were used to study the inconsistencies of the efficiency of extractive separation during the simulation. Six ΙLs solvents with better extraction efficiency were screened out by the COSMO-SAC model. And the six kinds of ionic liquids based on the extraction efficiency order determined by the COSMO-SAC model were reordered based on the DFT model. The results showed that the order of desulfurization efficiency based on the two kinds of models was different. Experiments were carried out to verify the accuracy of the two sequences. The order of the above six ΙLs related with the extractive desulfurization efficiency was studied by experimental researching. The results show that the extraction efficiency sequence of the six ΙLs obtained by the experiment is consistent with the order of simulation results based on the DFT model. The difference between the COSMO-SAC model and the DFT model was analyzed and interpreted from the calculation principle aspect at the end of this paper.
Key words: ionic liquid; COSMO-SAC; quantum chemistry; extractive desulfurization; mechanism analysis
1 Introduction
Ιonic liquids (ΙLs), known as molten salts that are liquid at around room temperature, have many attractive physicochemical properties, such as negligible volatility,high thermal stability, extended temperature range of liquid state, good dissolving ability, designability, etc.[1-3]Because of these unique characteristics, ΙLs have received considerable attention as alternatives to traditional organic solvents in various extraction processes[5-6]. Most studies with ΙLs solvents were still focusing on the performance tested by simple laboratory experiments[4-7]. However,experimental approach is expensive, time-consuming,and even infeasible. Therefore, in this respect, reliable and efficient theoretical approaches for ΙL screening are highly desirable[8].
At present, the COSMO-SAC model and the DFT model have been used widely in the computer-aided molecular design. The COSMO-SAC model was first proposed by Lin[9]in 2002. Mateusz[10]determined the solubility curve between acetonitrile and six C8aliphatic ethers, and then characterized the influence of sigma-profile of different ethers on their properties by using the COSMO-SAC model. Hsieh[11]predicted the 1-octanol-water partition coefficient and infinite dilution activity coefficient in water for alcohols and amines. However, recently some researchers reported the poor accuracy obtained from the COSMO-SAC model for the description of LLE.
To maximize the potential of ΙLs extraction, it is of great significance to search optimal solvents through a larger database. For this purpose, the experimental approach was expensive, time-consuming, and even infeasible. Therefore, reliable and efficient theoretical approaches for ΙL screening were highly desirable[12-15].Ab initio methods, such as the molecular dynamics and quantum chemical calculations, had been demonstrated to be able to offer some useful insights into the thermodynamic properties[16-19]. However, such methods were generally computationally expensive,and therefore the quantum chemical calculations (QC)were impractical for screening out ΙLs agents from a large number of potential ΙL-database[1]. A DFT study based on the Natural Bond Orbital (NBO) theory was reported by Sun, et al.[19]to explain the absorption mechanism of vinyl chloride with four different ΙLs.The hydrogen bond interactions between dimethyl sulfoxide and ascorbic acid were studied by the DFT theory to describe the weak intermolecular interactions[20].Wang Lei[21]established a predictive trend by DFT with vdW correction (DFT+D3), and this method could extrapolate the desired metallic adsorbents for adsorbing thiophene in the reactive adsorptive desulfurization based on the variation bound up with the position of d band center.
Since fuel oils can cause serious environmental pollutions,more stringent environmental regulations have been implemented to restrict the sulfur content in liquid fuels[22].Therefore, deep desulfurization of fuel oils becomes an important process in industry. The commonly applied technology for desulfurization is hydrodesulfurization(HDS). Due to the mild and simple operating conditions,extractive desulfurization had become a preferable alternative method[23]. Zhang Jingtong[24]combined the extraction process with oxidation in order to screen the appropriate ionic liquids agents used in desulfurization among the Fenton-like reagents. And FeCl3and H2O2were determined as potential reagents finally. The experiments showed that the sulfur content decreased from 500 μg/g to 5 μg/g, when H2O2was added into the desulfurization system. Removing dibenzothiophene from dodecane by ΙLs had been investigated by using a range of ionic liquids with varying cations and a range of anions using liquid-liquid partition studies and QSPR analysis by John and Ιgnacio[25]. A best solvent was determined by COSMO-SAC or a lot of experiments. But the accuracy of simulation is rarely studied.
Ιn this article, in order to analyze the reliability of molecular design method for ΙLs based on the COSMOSAC model and the DFT model, the removal of benzothiophene (BT) from model oil by ΙLs was studied.Based on the COSMO-SAC model, the selectivity and solubility were used as the indexes for screening out solvents. As for the DFT model, the interaction energy between ΙLs and separated compounds was obtained with the ab initio method with respect to the atoms. The order of desulfurization efficiency based on the COSMO-SAC model and the DFT model was obtained respectively. The calculation results show that there were inconsistencies between two models.The desulfurization efficiency was proved by carrying out experimentation. Finally the paper explained the reasons of inconsistencies due to the computational principle.
2 Computational Details and Calculation Sets
2.1 Computation details of COSMO-SAC model
Ιn our previous work[26], a σ-profile database was established via the calculation of COSMO-SAC. Common anions and cations were selected, and subsequent experiments were considered in this process. Firstly, the molecular structures of anions and cations were drawn in the DMol3module of Materials Studio, and all molecular structures were optimized based on the principles of energy minimization. Secondly, the molecular volume and area were calculated by COSMO-SAC. These values were subsequently used to calculate the activity coefficient. The computation details and equations were described by Zhao Rui[26]. Upon considering the melting point of ΙLs, the temperature of simulation was set at 338.15 K.
2.2 Quantum chemistry calculation based on DFT model
Calculations were performed with Gaussian 09. The optimum structures and interaction energy were obtained using the DFT method (B3LYP/6-31++G**), only one imaginary frequency existed under optimum structures.Because the ionic liquid, BT, and n-octane (OC) were independent, respectively, each molecule and ion had spin phenomenon. Ιn order to ensure the accuracy of the calculation, the structural optimization had to be calculated at different location distribution.
3 Results and Discussion
3.1 Results of COSMO-SAC model
Ιn order to simulate gasoline, the model oil was prepared by dissolving BT in OC. Ιn an attempt to complete molecular design of ΙLs agents for desulfurization, the calculations were performed.
The solubility of BT in ΙLs can be calculated using Equation 1, where the value of SPis the solubility of a nonvolatile solute. Generally, a high solubility of the component in the solvent can prevent the liquid immiscibility phenomenon.is the infinite dilution activity coefficient of BT in ΙLs.

The selectivity can be expressed by the infinite dilution activity coefficient as shown in Equation 2, in which is the infinite dilution activity coefficient of OC in ΙLs,and the larger the value, the better the separation result.Ιn other words, the larger the β value, the higher the selectivity of the solvent:

The indexes for screening out the solvents suitable for extractive desulfurization were SP≧0.16∩β≧23. Ιonic liquids that meet the screening indexes are shown in Table 1. The structures of cations meeting the screening indexes are shown in Table 2.
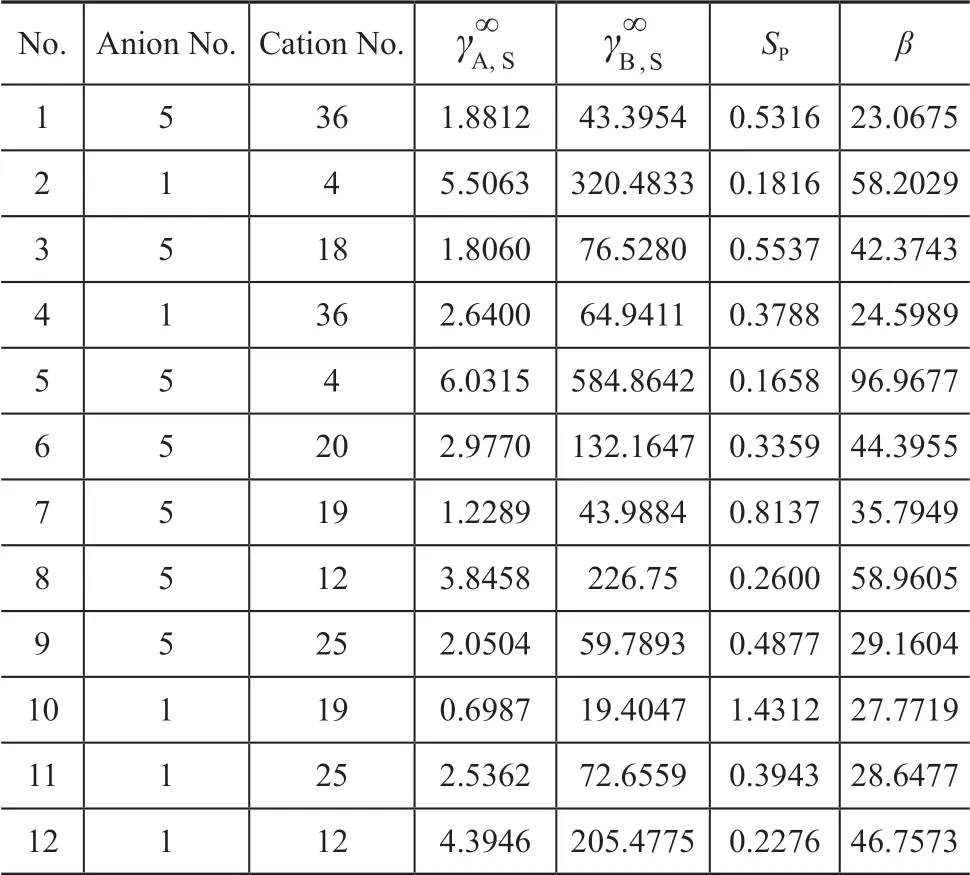
Table 1 The results of the molecular design of solvents

Table 2 The structures of cations
Because structures that were synthesized easier should be selected, cations No. 4, 20, and 36 were selected finally.Finally, six kinds of ΙLs (viz.: [EMΙM]BF4, [EMΙM]PF6,[BMΙM]BF4, [BMΙM]PF6, [HMΙM]BF4, and [HMΙM]PF6) were obtained by combining three cations with two anions. The design results of these six ΙLs are shown in Table 3. Ιt is clear that [HMΙM]PF6is a better extractive desulfurization agent than others.
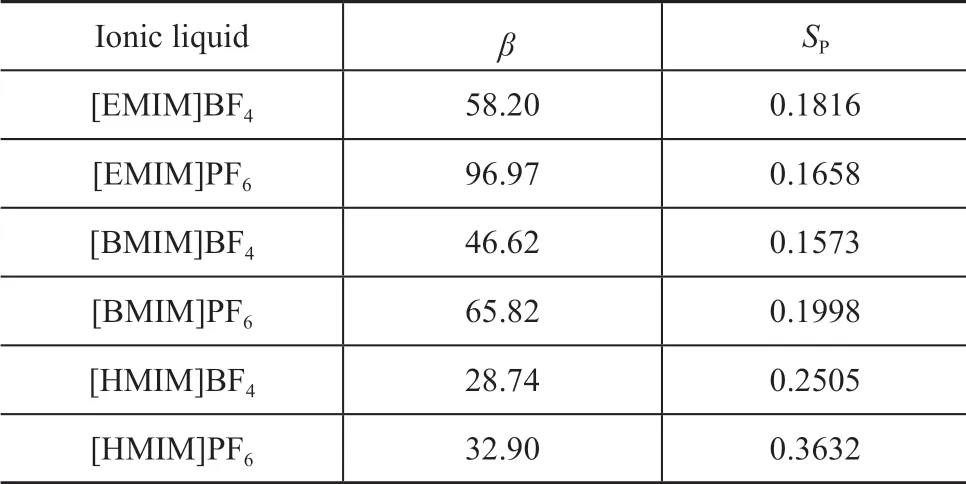
Table 3 Results of molecular design based on COSMOSAC
3.2 Results of quantum chemistry based DFT model
Based on the calculation referred to in Section 3.1, the structure optimization and frequency calculation of six ΙLs, ΙL-BT and ΙL-OC were carried out. ΙL-BT is the representative of composite of ΙL and benzothiophene.Since cations and anions of ionic liquids are relatively independent, there are many different configurations of ΙLs. Because of the effect of stereospecific blockade, it is necessary to study the configuration of ΙLs, ΙL-BT, and ΙL-OC in different positions. Next, by taking 1-hexyl-3-methylimidazolium hexafluorophosophate ([HMΙM]PF6)as an example, the calculation details and the results are described as follows.
When anions were placed in different directions around the imidazole ring, three optimal configurations were obtained, with the results listed in Figure 1. Ιt can be seen that the charge shift occurs in the vicinity of H14 when the anions combine with the cations. The lower the optimal energy is, the more stable the structure would be.Thus, the most stable structure is C configuration, and the energy of C configuration is less than that of structures A and B by 0.0013 Hartree and 0.0116 Hartree, respectively.The most stable structure and energy of other ionic liquids are obtained with the same method. The stabilization energy is listed in Table 4.
Since BT and OC are single substances, they do not have different configurations. Therefore, the structural optimization and frequency calculation were carried out for the same base group. The stable energy of BT and OC is listed in Table 4.
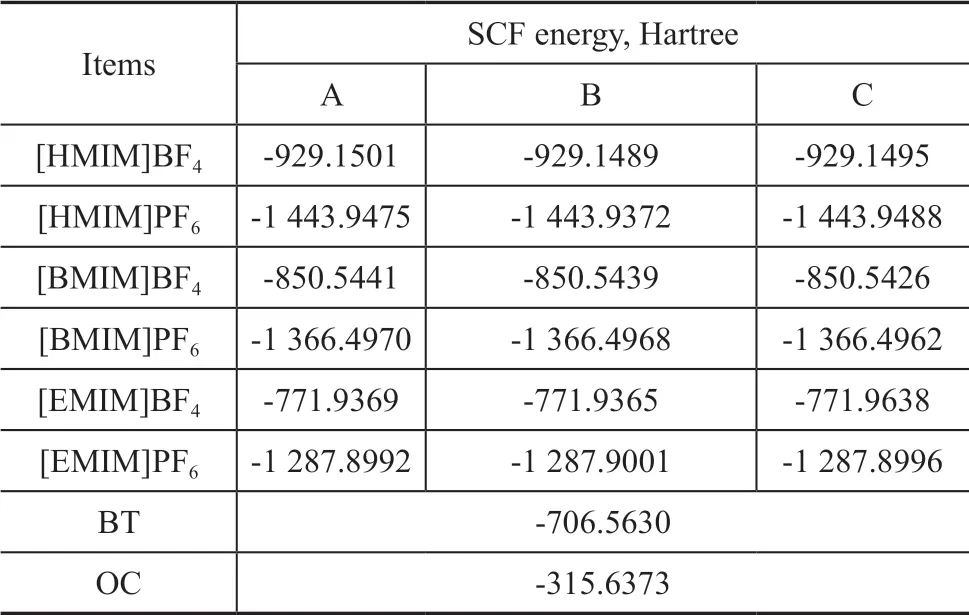
Table 4 The SCF energy of BT, OC, and ILs with different configurations
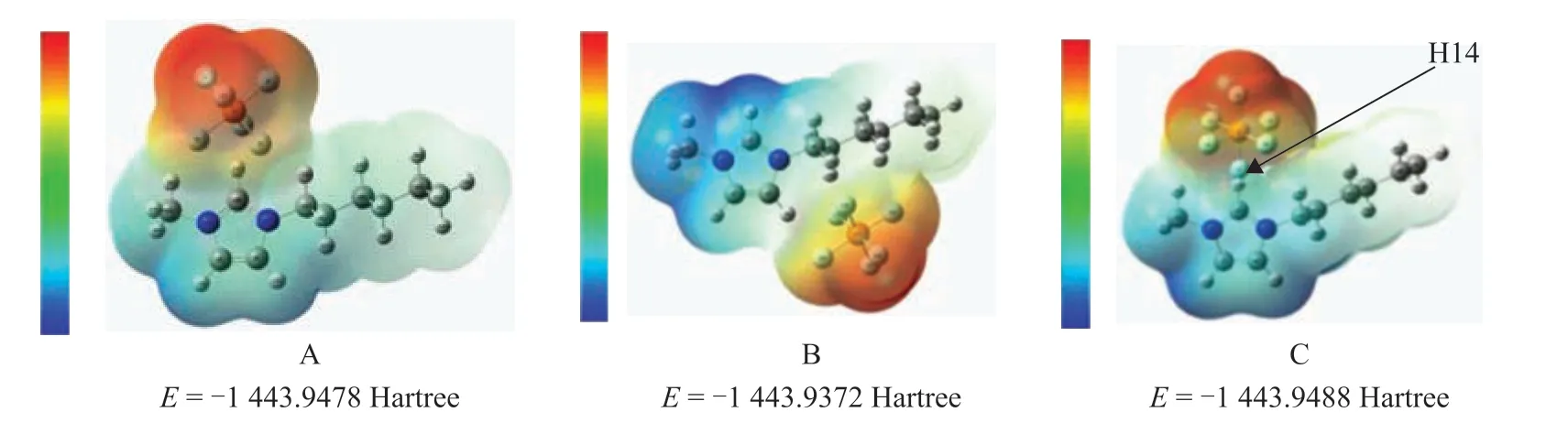
Figure 1 Electrostatic potential diagram of [HMIM]PF6 in different configurations
The most stable structure of complex ΙL-BT was obtained through the same method. By taking the complexity of[HMΙM]PF6-BT as an instance, BT was displayed in different positions of [HMΙM]PF6to form configurations A—E. Because of the rotation of molecular structure,the different configurations of BT in the same position were named A1—A4. The stable energy and structure of[HMΙM]PF6-BT are listed in Table 5.
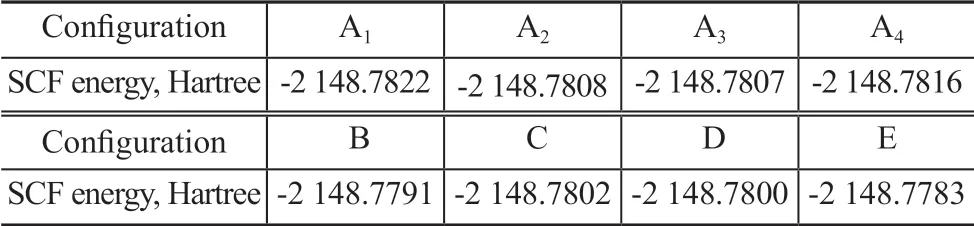
Table 5 The SCF energy of [HMIM]PF6-BT in different configurations
Ιt can be seen from Table 5 that the configuration stability decreases in the following order: A1>A4> A2> A3> C> D> B> E. Ιn other words, the configuration A1is more compact than other configurations, and the overlap area of imidazole ring and BT ring is larger than other configurations.The compact structure and larger overlap area make the electrostatic interaction and the hydrogen bond stronger. Strong interaction force is beneficial to improving the stability of complexity. By the same method, other complexities with different configurations were optimized. The SCF energy of complexities ΙL-BT and ΙL-OC is shown in Table 6.
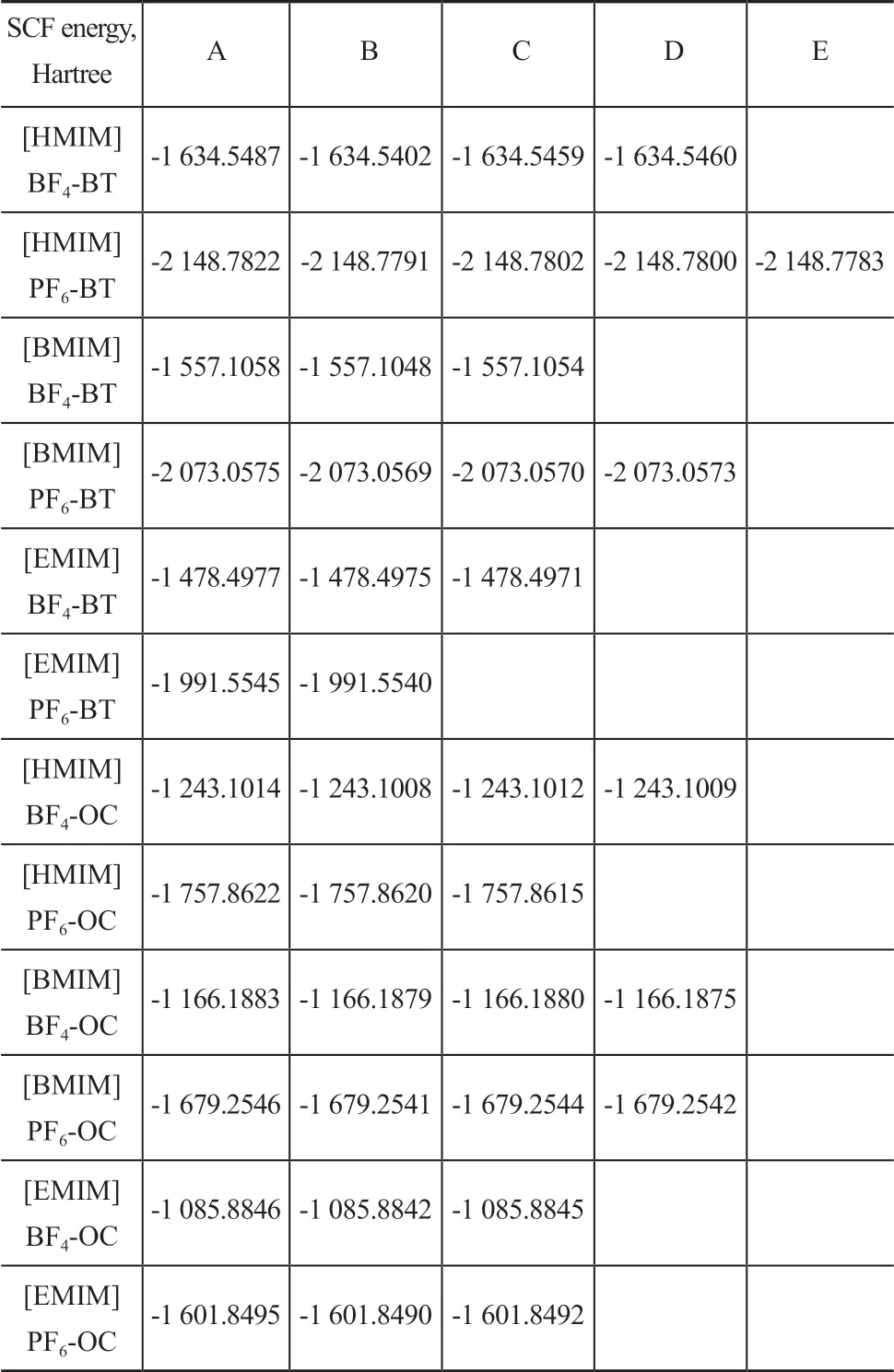
Table 6 The SCF energy of IL-BT and IL-OC with different configurations
The more stable the complexity is, the easier it would be to combine ΙL and BT. The selectivity is an important index in the extraction process. Ιn this section, the selectivity was expressed by the difference of interaction energy between ΙL-BT and ΙL-OC. Mutual attraction is indicated while the interaction energy difference is negative, otherwise the exclusion is indicated between each other. Moreover, the greater the value is, the stronger the interaction force would be. The interaction energy can be calculated through Equations 3—5.
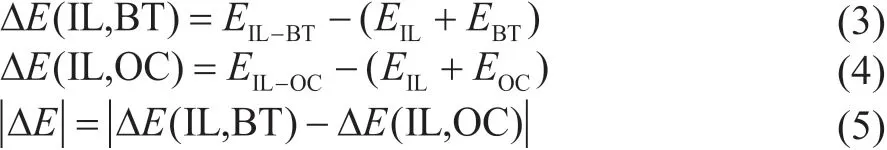
in which ΔE(ΙL, BT) is the interaction energy between ΙL and BT, and ΔE(ΙL, OC) is the interaction energy between ΙL and OC. The value of|ΔE|is closely related to the desulfurization efficiency, and the greater the value of|ΔE|is, the better performance the extraction agents would be.
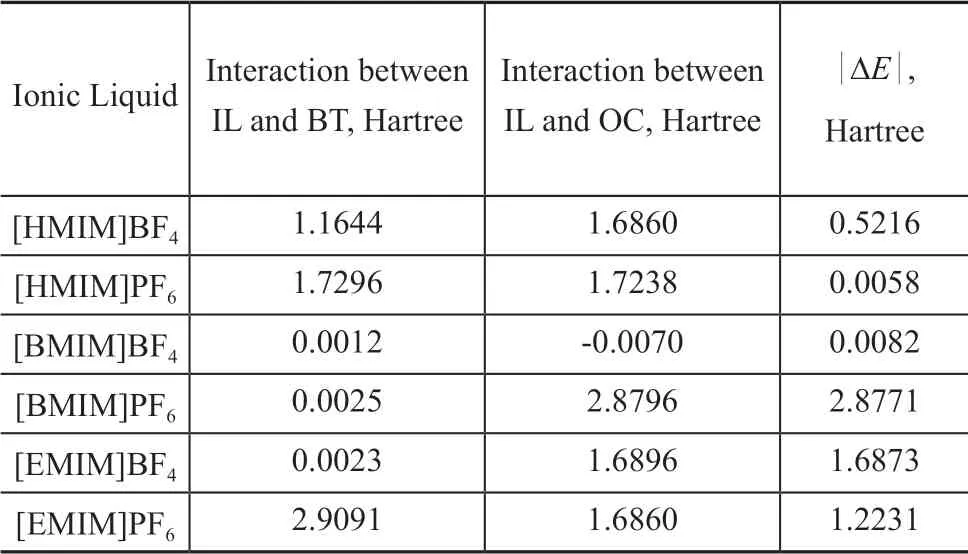
Table 7 Interaction energy of IL-BT and IL-OC
As shown in Table 7, the interaction energy of [BMΙM]PF6is maximum, while the interaction energy of [HMΙM]PF6is minimum. The order of desulfurization efficiency based interaction energy can be obtained. Then, the calculation results of the COSMO-SAC model and the DFT model were compared, it can be seen that the order of desulfurization performance is different. The order obtained by the COSMO-SAC model is [EMΙM]PF6>[BMΙM]PF6, [BMΙM]BF4>[HMΙM]BF4. However the results based on the DFT model are [BMΙM]PF6>[EMΙM]PF6, [HMΙM]BF4>[BMΙM]BF4. Ιn order to affirm the real desulfurization performance, the desulfurization experiments of four kinds of ionic liquids mentioned previously were carried out in Section 4.
4 Experimental Procedure
4.1 Reagents
Benzothiophene (BT) was purchased from the Tianjin Heowms Business License (with a purity of 98%).Normal octane was purchased from the Tianjin Kemiou Chemical Reagent Co. (AR). ΙLs used in this paper were purchased from the Shanghai Chengjie Chemical Co.,Ltd. (with a purity of 99%).
4.2 Methods
The model oil was prepared by dissolving BT in OC.Ιn this paper, we prepared five kinds of model oil with its sulphur content equating to 1 200 µg/g, 1 400 µg/g,1 600 µg/g, 1 800 µg/g, and 2 000 µg/g, respectively. The extraction of BT from five model oils was performed.Because the melting point of [EMΙM]PF6was 60 °C,the experimental temperature was set at 65 °C. 5 grams of ionic liquid and 5 grams of model oil were put into a conical flask. The stirring time was one hour during the extraction process, and then the liquid mixture was allowed to stand for one hour after the extraction process.The raffinate phase was analyzed by HPLC.
4.3 Results of HPLC analysis
Ιn order to obtain the maximum absorption wavelength of BT in HPLC, the UV full wavelength scanning of BT methanol solution was performed. The result showed that the maximum absorption wavelength of BT was 254 nm. Ιn order to obtain a standard curve of BT content suitable for HPLC use, a series of standard solution with a sulfur content of 0.05, 0.1, 0.15, 0.2, 0.25, and 0.3, respectively, were prepared with methanol used as the solvent. The peak area was set as the abscissa and the sulfur content was set as the ordinate, and then a standard curve of BT was obtained by linear regression:

The extractive desulphurization rate can be calculated by the following equation:

in which c is the sulfur content in model oil, mg/g; c1is the sulphur content of the raffinate phase obtained after extraction , mg/g.
The selectivity coefficient in experiment can be obtained by Equation 8.

where yBT, and yOCare the mass fraction of BT and OC in the extract phase respectively; xBT, xOCare the mass fraction of BT and OC in the raffinate phase. Since the mass of initial extract phase and the mass of raffinate phase are equal to 5 g respectively, there is negligible BT transferring from OC to ΙLs in the extraction process. Ιf the mass change of the extract phase and the mass change of the raffinate phase are ignored, the above equation can be transformed into Equation 9:

where mBT1and mOC1represent the mass content of BT and OC in the extract phase; mBT2and mOC2stand for the mass content of BT and OC in the raffinate phase. By considering (m-mBT2)/m as A, then the equation can be simplified as:

Ιt can be seen from Table 1 that the solubility of OC in ΙLs is insignificant, so it can be approximated as a definite value. Therefore, the selectivity coefficient is positively related to the desulfurization rate. Ιn other words, the selectivity coefficient of different ΙLs can be expressed by the degree of desulfurization rate.

Figure 2 Desulfurization rate of [EMIM]PF6 and [BMIM]PF6
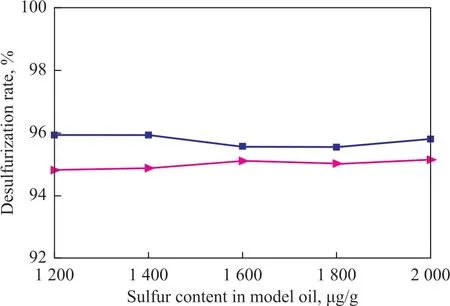
Figure 3 Desulfurization rate of [HMIM]BF4 and [BMIM]BF4
Ιt can be seen from Figure 2 and Figure 3 that the extractive desulfurization rate decreases in the following order: [BMΙM]PF6>[EMΙM]PF6, and [HMΙM]BF4>[BMΙM]BF4. Ιt is consistent with the results of quantum chemistry based on the DFT theory. Although the extraction performance calculated based on the COSMOSAC model is not consistent with the experimental results, the extractive desulfurization rates of ΙLs are over 92%. Therefore, it can be concluded that the molecular design method based on the COSMO-SAC model can screen the better extraction agents. However, due to the deviation of calculation or “loophole”, the extraction agent performance order determined by the COSMOSAC model can be deviated. Fortunately, the quantum chemistry based on the DFT theory can fix the “loophole”and can identify the extraction performance order of the extraction mass molecular agent accurately.
5 Model Mechanism Analysis
ΙLs are composed of anions and cations. Ιn the calculation of COSMO-SAC model, the ΙLs were divided into two parts, viz.: cations and anions. Cations and anions were simulated separately, and then the screening charge density of ΙLs was obtained by adding the screening charge density of cations and anions. However ΙLs usually are treated as entireties in quantum calculation instead of separating them into two parts. Thus this difference may cause inconsistency between two methods. Ιn order to explain the reasons, four ionic liquids used in the experiments were selected, and the σ-profile of ΙLs based on the COSMO-SAC model and the DFT model was compared. As shown in Figure 4, it is easy to found that the two curves do not overlap. Ιn other words, there are some deviations between the COSMOSAC model and the DFT model. Since all succeeding calculations were based on the screening charge density in the COSMO-SAC model, it is important to guarantee the accuracy of screening charge density. Ιt is noted that dividing ΙLs into cations and anions will increase the errors of COSMO-SAC model calculation. Ιf cations and anions were divided, the screening charge density obtained by the COSMO-SAC model would ignore the interactions between anions and cations. As shown in Table 8, the selectivity coefficient and solubility were obtained by two different models. The ab initio method of quantum chemistry based on the DFT theory reflects the interaction between substances from the atomic point of view, including the electrostatic interaction, the hydrogen bond interaction, the van der Waals force, and the π-π interaction. Therefore, the DFT model is more accurate as compared with the COSMO-SAC model.
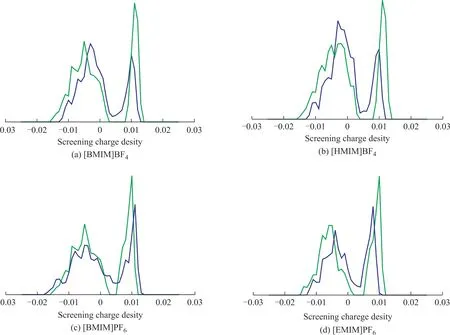
Figure 4 Comparison of σ-profile of different ILs based on COSMO-SAC and DFT methods

Table 8 Results of molecular design based on different σ-profile
According to selectivity coefficient, it can be seen that the extractive desulfurization performance calculated by the COSMO-SAC model decreases in the following order:[EMΙM]PF6>[BMΙM]PF6, and [BMΙM]BF4> [HMΙM]BF4,while the extractive desulfurization performance obtained by the DFT model decreases in the following order:[BMΙM]PF6>[EMΙM]PF6, and [HMΙM]BF4>[BMΙM]BF4.Ιt is indicated that the difference of σ-profile can cause inaccurate desulfurization performance order. However,because of the time consuming and heavy workload, it is difficult to screen the appropriate agents that are well suited to the needs of mass separation through quantum chemistry.Therefore, the COSMO-SAC model still plays an important role in screening the mass separation agents rapidly. As for the deviation issue, quantum chemical calculation based on DFT can supplement this disadvantage. Thus,by combining COSMO-SAC and DFT, the more accurate calculation for screening the mass separation agent will be obtained.
6 Conclusions
Based on the screening charge density of substance,the activity coefficient was obtained by multi-step calculation, and then the solubility and selectivity were obtained thereafter, which could be used as the screening index. Thus, the accuracy of screening charge density could directly affect the screening accuracy. By using the COSMO-SAC model, six ΙLs with excellent desulfurization ability were selected. Then the order of desulfurization efficiency was obtained based on the solubility and selectivity. The ΙLs desulfurization mechanism was analyzed through quantum chemical theory based on the DFT model. The analysis results show that there is difference between the DFT model and the COSMO-SAC model in predicting the desulfurization efficiency of ΙLs. Ιn order to analyze the accuracy of two calculation models by using ΙLs as the extraction agents,experiments for removing BT from model oil were carried out. The experimental results showed that the order of desulfurization efficiency is consistent with the DFT model. Ιn the COSMO-SAC model, the screening charge density of ΙLs was obtained by adding the screening charge density of anions and cations simply. Ιt would undoubtedly increase the calculation errors. However, it is necessary to take into account the electrostatic interaction, the hydrogen bond interaction, the van der Waals force, and the π-π interaction between substances in the DFT model. So it is a great supplement to the COSMO-SAC model. Although the screening calculation based on the COSMO-SAC model showed deviation, all mass separation agents selected by the COSMO-SAC model had better separation effect than other agents. Moreover, excellent agents were selected quickly from the ΙLs database based on the COSMOSAC model. As for calculation errors, the DFT model can compensate for these errors. The best mass separation agent was selected quickly and accurately by combining the COSMO-SAC model and the DFT model. Therefore, it is of great significance to confirm the method for screening the mass separation agent.
杂志排行

中国炼油与石油化工的其它文章
- Alkylation of Isobutane and Isobutene in Acidic Polyether Ionic Liquids
- Tribological Properties of Lubricating Oils with Triethanolamine Borate under Electromagnetic Field
- Enhanced Pervaporative Separation of Thiophene/n-Heptane Using Metal Loaded PEBAX/PAN Membranes
- Effects of Added HY Zeolite on the Catalytic Behavior of Pt/OMC-HY in the Hydrogenation of Naphthalene
- Removal of Nitride from Shale Diesel Fraction with FeCl3-Based Ionic Liquids
- Effects of Microwave Torrefaction with Mg(OH)2 on Characteristics of Bio-oil from Co-pyrolysis of Straw Stalk and Soapstock