基于全血基因表达谱的脑组织基因表达量预测模型的建立
2019-10-24徐文剑
徐文剑 李 巍
(国家儿童医学中心 首都医科大学附属北京儿童医院 遗传与出生缺陷防治中心;北京市儿科研究所 出生缺陷遗传学研究北京市重点实验室;儿科重大疾病研究教育部重点实验室,北京 100045)
基因表达实验通过刻画细胞转录水平变化情况来阐释生物学表型,是辅助疾病机利解析、药效评价和临床疾病亚型分类等工作的有力工具[1-5]。关于转录组测序和基因芯片表达谱的基因表达综合库(gene expression omnibus,GEO)、基于网络的细胞反应印记整合图书馆(library of integrated network-based cellular signatures,LINCS)和基因型-组织表达(genotype-tissue expression,GTEx)项目等公共数据库和基因表达数据分析算法应运而生[6-9]。随着实验分析成本下降和组学数据分析流程完善,基于转录组测序的基因表达谱分析最终将从基础研究走向临床应用。
疾病通常有明确的受累组织,需要采集疾病相关的组织样本,使用基于转录组测序或微阵列基因表达谱分析实验以便精准辨识出疾病相关组织细胞内中基因异常表达情况。例如孤独症谱系障碍(autism spectrum disorder,ASD)的遗传学分子机制可以通过基因表达分析来辅助解析,常用组织包括全血、淋巴母细胞系、小脑、额叶皮质、前额叶皮质、颞叶皮质、尾状核等[10-12]。最近Quesnel-Vallières等[13]报道至少有12篇文献采用前额叶皮质、颞上皮质和小脑等脑区组织样本来进行基因表达分析来研究ASD遗传基因作用机制和相关基因表达标志物。
脑组织样品的基因表达实验样品收集工作难度大、风险高、成本高,限制了组织特异性基因表达谱分析在该领域的发展,因此亟需一种能规避脑组织取样的表达谱检测替代方案。全血样本是一个无创、易采集的替代性组织,并且与脑组织有基因表达模式的相似性。例如Mundalil Vasu等[14]报道血清中的一组microRNA(miRNA)分子有可能成为ASD的非侵入性生物标志物。Sullivan等[15]报道了基因在血液与17个大脑组织的表达量成对斯皮尔曼(Spearman)相关系数中位数为0.5,血液与中枢神经系统组织有中等强度的相关性。Bosker等[16]报道了精神分裂症(schizophrenia)动物模型在不给药且不恐惧刺激条件下前扣带回皮质和血液白细胞的基因表达量有弱相关性,在给药并且恐惧刺激条件下有强相关性。Hensman等[17]利用亨廷顿症(Huntington’s disease,HD)的两个人群队列中血液样本,发现这两组HD患者血液与公共数据库中HD患者尾状核的基因表达模式有显著的一致性。
从血液组织表达数据推断另一个组织中的基因表达量已被证明是可行的。Halloran等[18]曾报道利用血液与肺组织的基因表达的相关性建立过肺组织基因表达量预测模型,使用GTEx项目31对全血和肺组织样本转录组数据为每个基因分别构建了以年龄、性别、该基因的血液表达量为输入变量预测该基因的肺组织表达量的线性回归模型,结果显示18%的基因的血液表达量与肺组织表达量显著相关。本研究立足于全血转录组基因表达量与脑组织中基因表达量的潜在相关性,挖掘了基因表达量跨组织多对多关系,构建了一个基于全血转录组表达量数据的未取样脑组织中基因表达量的计算预测模型。
1 材料与方法
1.1 数据来源及处理
1)数据下载:截至2019年4月,GTEx研究联盟收集并以高通量转录组测序(RNAseq)研究了来自714名生前健康的人类捐献者的11 688份尸检组织样本,涵盖53个组织,包括实体器官组织、脑分区、全血、两种来自血液和皮肤的细胞系,这些样本为研究基因表达的组织特异性和个体特异性提供了关键材料。该项目产生的所有分析数据保存在公共数据库GTEx网站上,每位捐献者的一个生物组织样本对应着表达量数据集中的一条记录。
从GTEx数据库(https://gtexportal.org/)下载相关RNAseq基因原始表达量数据文件,主要包括2016-01-15_v7版本的每百万读序列的单位长度转录本(transcripts per kilobase million,TPM)表达量文件(GTEx_Analysis_2016-01-15_v7_RNASeQCv1.1.8_gene_tpm.gct.gz,841M)和GTEx数据库样本属性的注释信息(GTEx_v7_Annotations_SampleAttributesDS.txt,7.9M)。
2)数据分割:本研究期望建立的每个脑组织基因表达量预测的模型,要求同一个组织捐献者同时有全血样本和脑组织基因表达量数据。分别为每个脑组织从原始数据文件gct中提取出全血样本与靶组织样本的捐献者编号(subject ID)完全匹配的数据条目,分别命名为靶组织基因表达量预测任务对应的全血表达量特征数据集(例如Brain-Cortex_features.txt)和靶组织基因表达量目标数据集(例如Brain-Cortex_targets.txt),二者的数据条目一一对应,是待构建模型的实例数据。
3)数据清洗:在匹配全血表达量特征数据集中的每个基因表达量特征在部分捐献者条目存在TPM表达量为 0的情况,不能区分是测序建库的误差还是样本确实未表达该基因。为减小数据噪声,剔除存在表达量TPM为0情况的特征,只保留所有捐献者条目中表达量值完整的特征。同理,对靶组织基因目标数据集进行处理。各预测任务的特征维度在14 000左右,目标维度在19 000左右。
4)数据标准化:各组织表达量子数据集为二维矩阵,一个维度代表样本,另一个维度代表基因。基因的表达量差异有统计学意义,表达量较高和较低的基因表达量数值的平均值可相差若干个数量级,无法直接用于回归建模。因此使用python sklearn模块中preprocessing.StandardScaler()函数依次在每个基因的维度上进行样本表达量数据的Z-score标准化,得到均值为0,标准差为1的新的表达量值矩阵,使得所有基因的表达量达到同一个数量级且保留样本间的差异信息。
1.2 基于线性模型的特征选择
本研究构建基于最小二乘回归(least squares regression, LSR)的线性回归预测模型。本研究的样本量区间为[50,101],特征数均为104数量级。本研究采用经验法则:要求特征维度必须小于样本量,样本量是特征数4倍及以上。弹性网络(elastic net)是使用L1,L2范数作为正则项的线性模型,能学习出只有少量非零系数的稀疏模型,作为特征选择的方法已报道[19-21]可应用到基于生物医学文本的重症监护病房(intensive care unit,ICU)的患者病情分级、基于基因表达谱和基因变异信息的药物敏感性预测、脑区静息态功能性磁共振成像[resting state functional magnetic resonance imaging,(rs)fMRI]等相关领域,因此本研究选用弹性网络作为特征选择方法。
预实验阶段先以基于弹性网络模型的特征选择方法分别为每个脑组织的前200个基因预测任务提出5、10、15和20个最相关的全血基因表达量特征,调用函数为sklearn模块中的linear_model.ElasticNet(alpha=0.02, max_iter=10 000)。之后综合考虑预测模型性能和可用样本量的限制条件,从4种方案中选择最佳关键特征个数完成剩余1万多个基因预测任务的特征选择。
以一个特定脑组织为例,对建模过程予以说明。设预处理后的全血表达量数据矩阵为Dblood,样本量为S,共有M个基因的表达量信息,样本i的全血表达谱数据记为向量xi=(xi(1),xi(2),…,xix(m))其中i∈(0,S),该脑组织表达谱数据矩阵为Dbrain,样本量同为S,共有N个基因,样本i的脑组织表达谱设为向量yi=(yi(1),yi(2),…,yix(N))其中i∈(0,S),针对以xi为自变量预测靶基因t的表达量yi(t)的基本预测任务,在特征选择阶段,选取弹性网络模型为基因t的目标函数,求解最优参数
(1)
其中模型参数w(t)∈RM,b(t)∈R,正则项比例系数a∈(0,1)。

1.3 基于全血基因表达量的多组织基因表达量预测模型构建
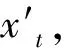
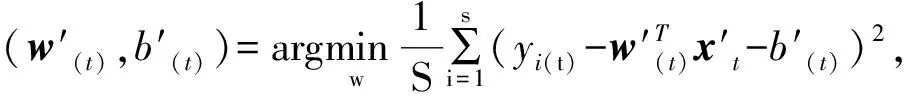
(2)
1.4 统计学方法
1.4.1 预测模型性能评估指标

1.4.2 预测值与真实值的决定系数
2 结 果
2.1 脑组织与全血组织配对的可用样本量统计
GTEx表达量数据集中包含13个脑组织,根据捐献者样本编号将全血样本和脑组织样本进行配对,经数据分割、清洗、标准化,分别将全血、每个脑组织的表达量数据提取为单独数据文件。统计可用样本量和可用基因个数,详见表 1。
2.2 脑组织不同特征选择方案下前200个基因表达量预测模型性能
各脑组织与全血配对可用的样本量最小只有50。为确定特征选择方案,将前200个基因预测任务作为预实验。每个脑组织的前200个基因表达量预测任务分别提取5、10、15和20个最相关的全血基因表达量特征,基于全血基因表达量的LSR脑组织基因表达量预测模型的MAE结果汇总详见表2。

表1 GTEx数据集中脑组织与全血样本配对可用样本统计信息Tab.1 Summary of available sample of brain tissue matched with blood in GTEx dataset
GTEx:genotype-tissue expression;BA:brodmann area; .
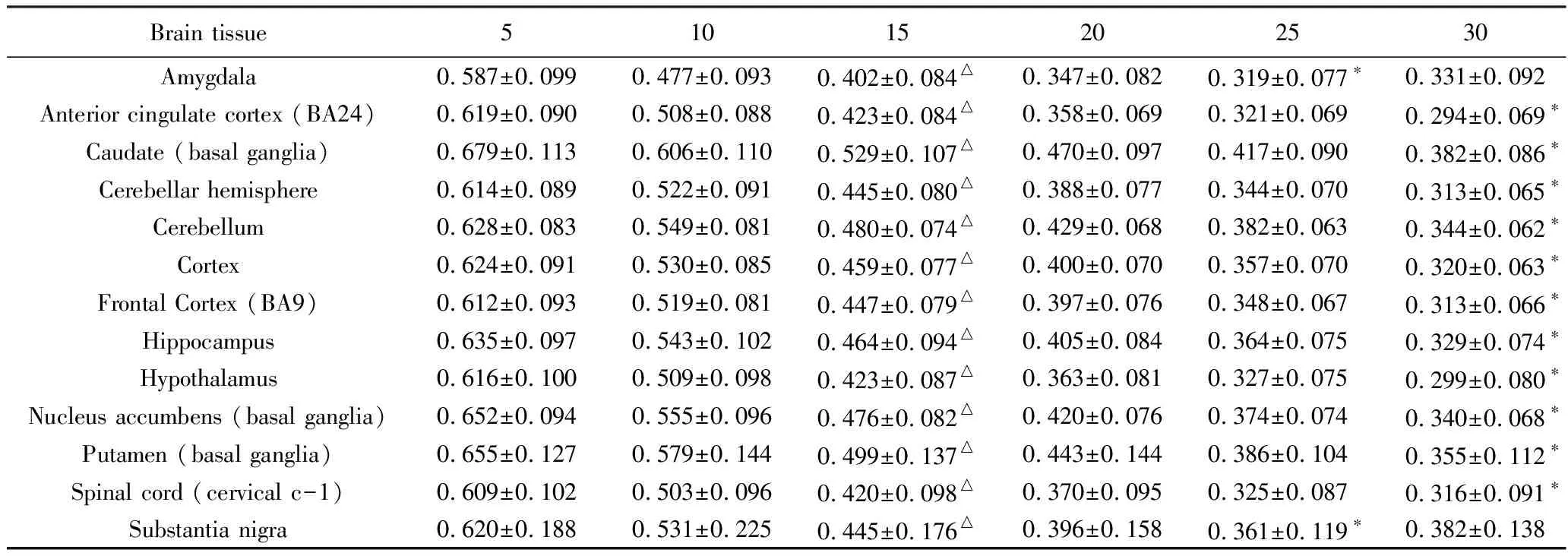
表2 各脑组织前200个基因表达量预测模型平均绝对误差Tab.2 MAE of first 200 gene expression prediction task of each brain tissue
*: MAE of optimal feature sets;△: MAE of used feature sets;MAE:mean absolute error;BA:brodmann area.
对于杏仁核(amygdala)和黑质(substantia nigra)两个样本量最小(n=50,表 1)脑组织,提取25个最相关特征的预测模型达到最优预测性能,提取少于25个特征处在欠拟合区域,提取多于25个特征则处在过拟合区域;其他10个脑组织在30个最相关特征时达到最优性能(表 2中以*表示),仍处于欠拟合区域。为保证脑组织最小样本量是特征数的4倍左右,统一选择提取15个最相关特征的方案(表2中以Δ表示)。重新用全部样本训练线性回归模型,此线性模型得到的脑组织前200个基因表达量的预测值与真实值的拟合度良好(图 1)。
2.3 脑组织全基因表达谱预测模型性能
根据预实验结果,提取包含15个最相关的全血基因表达量特征构成低维度新特征数据集,并构建13个脑组织所有基因表达量线性回归预测模型,预测模型在交叉验证集上的性能总结详见表3。所得的一系列线性回归预测模型由式子(2)中最优参数表示。预测模型MAE和RMSE结果趋势一致。其中杏仁核(amygdala)组织预测性能最佳,尾状核(caudate (basal ganglia))组织预测性能最差。
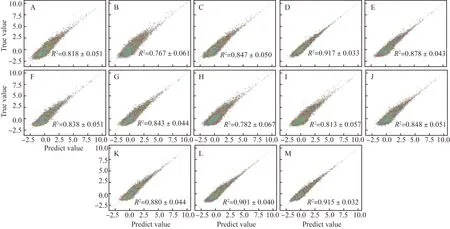
图1 13个脑组织前200个基因表达量预测值与真实值的线性拟合度Fig.1 Goodness of fit of model predicted value and true value of 200 genes in 13 tissues
A:cortex;B:cerebellum;C:hippocampus;D:substantia nigra;E:anterior cingulate cortex;F:frontal cortex;G:cerebellar hemisphere;H:caudate (basal ganglia);I:nucleus accumbens (basal ganglia);J:putamen (basal ganglia);K:hypothalamus;L:spinal cord (cervical c-1);M:amygdala.
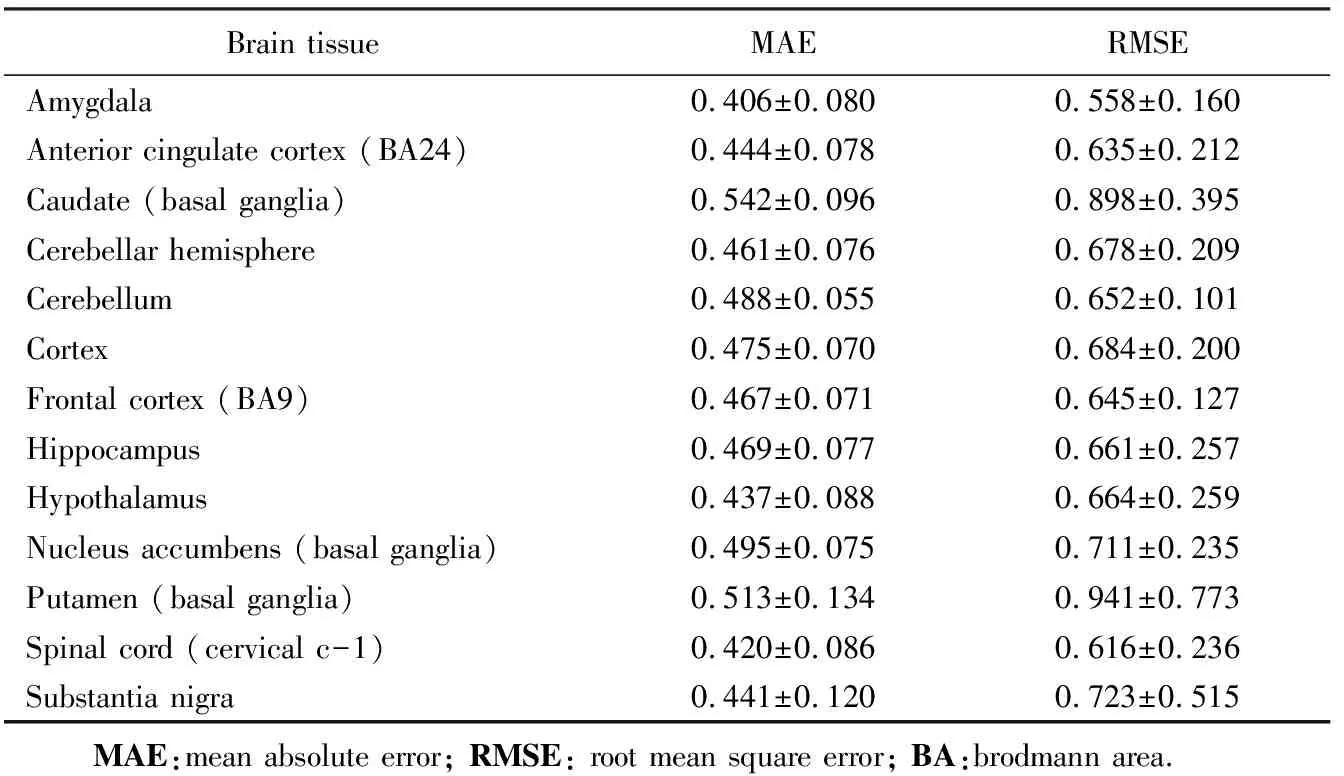
表3 各脑组织基因表达量预测模型平均绝对误差和均方根误差Tab.3 MAE and RMSE of gene expression prediction model of brain tissues
3 讨论
脑组织样本手术取样限制了相关组织内基因表达分析的大规模开展。本研究针对临床研究中的脑组织样本收集困难这一实际问题,从GTEx转录组数据集中挖掘脑组织转录组与全血转录组的内在数值相关性,期望摸索出一个利用有限样本集构建具有一定准确度的转录组预测模型的通用设计模式。相比组织转录组的稳定性,全血转录组信息动态变化速度较快。受到GTEx数据集样本量的规模所限制,从全血转录组中能够间接推测出组织转录组真实信息的比例及线性预测模型可靠性依然是未知数。采用特征选择方法对每个单元预测任务进行高维特征预处理,一是为提取出有利于提高预测性能的关键特征,二是起到过滤全血转录组中无关基因表达数据的干扰。
本研究应用常规线性回归模型建立了基于全血转录组表达量数据的脑组织基因表达量的一组预测模型,表明仅用全血表达谱数据能比较准确地预测出未取样脑组织基因表达量,将来进一步发展成熟后,或许可以在转录组研究中规避脑组织样本的手术取样。除了孤独症谱系障碍,脑组织基因表达量预测模型也能为帕金森病和精神分裂症、双相情感障碍、抑郁症和酗酒等重要精神障碍疾病的基因表达谱研究提供一种备选工具[22-27]。
本研究的总体目标是针对每个靶组织独立地构建一个基于全血样本基因表达量的组织内基因表达量回归预测模型,预测模型的输入特征为个人全血样本基因表达量,预测值为靶组织内各基因表达量。回归预测模型性能评估环节计算预测表达量与真实表达量的差异程度。将一个靶组织预测模型构建任务化整为零,分解为靶组织所有基因表达量预测和靶组织的单基因表达量预测模型两个层次。预测模型的最小单元任务为基于全血基因表达量数据的靶组织中的单基因表达量预测模型构建问题。
全血表达量数据包含约104个转录本,平均每个组织只有少于100个数据样本,可知输入特征的维度将高出样本量2个数量级,原输入特征直接用于回归建模会带来过拟合问题。因此本研究用特征选择方法计算出每个输入特征与预测目标的关联程度表征数值,经排序选择出适合训练集样本量规模和预测问题性质的一组最优的输入特征。生物医学数据集通常具有特征维度高和样本量小的特点,阻碍了挖掘其中所蕴含的海量生物学信息和解码深层次基因功能的基础数据研究,特征提取日益成为生物医学大数据发展和应用的一种专门技术[28]。本研究初步研究了应对这一问题的一种生物信息学分析思路,本预测模型为组织转录组分析建模工作提供了新的视野。
