HPLC 法测定青蒿素哌喹片中哌喹有关物质
2019-10-22张志坚杨兆丽张加颂谢丹娥
张志坚 杨兆丽 张加颂 刘 澎 谢丹娥 王 琪
青蒿素哌喹片为青蒿素和哌喹组成的复方制剂,2006 年拿到国家一类新药证书,具有自主知识产权,是广东新南方青蒿药业有限公司独家生产品种。该药主要用于治疗恶性疟、间日疟和三日疟,在国外尼日利亚、肯尼亚、坦桑尼亚等20 多个国家获得注册批件和专利,青蒿素哌喹片原标准为YBH08022006[1],现行标准为中国药典2015 年版二部“青蒿素哌喹片”项下标准[2],采用高效液相色谱(HPLC)法检查有关物质,但该法检测灵敏度低,0.05%自身对照溶液几乎不能检出,检测波长选用216nm 不合适,检测结果准确度低,且检测结果偏高,与实际情况不符。为严格控制药品质量,我单位决定优化质量标准,提高制剂中哌喹有关物质的灵敏度、专属性和可靠性,本文参考2015 版中国药典中“磷酸哌喹”[3]及其他文献[3-8]中有关哌喹、磷酸哌喹类有关物质的测定方法,研究HPLC 法测定青蒿素哌喹片中哌喹有关物质的方法,并建立哌喹有关物质限度[2-9]。
1 仪器与试药
1.1 仪 器 安捷伦1100 高效液相色谱仪;赛默飞Ultimate3000 高效液相色谱仪;色谱柱:Thermo BDS HYPERSIL C18 柱(250mm×4.6mm,5μm)、YMC-Triart C18 柱(250mm×4.6mm,5μm);XP204S 型分析天平(梅特勒-托利多有限公司)(1/万);XS205DV 型分析天平(梅特勒-托利多有限公司)(1/10 万);FE20 型PH 计(梅特勒-托利多有限公司)。
1.2 试 药 青蒿素哌喹片,广东新南方青蒿药业有限公司,批号20141001、20141002、20 141003;哌喹,广东新南方青蒿药业有限公司,批号20 161030;青蒿素,广东新南方青蒿药业有限公司,批号Y13 120001;杂质I[7-氯-4-(1-哌嗪基)喹啉],中国药品检定研究院,批号101339- 201501;杂质II(7-氯-4-羟基喹啉),中国药品检定研究院,批号101340- 201501;杂质Ⅲ[1,4-二(7-氯喹啉-4-基)哌嗪],中国药品检定研究院,批号101341- 201501;甲醇、乙腈均为色谱级(上海安谱);水为超纯水;三氯乙酸(分析纯,国药集团);三乙胺(分析纯,国药集团);磷酸(分析纯,国药集团);其余试剂均为分析纯。
2 方法与结果
2.1 色谱条件及系统适用性试验 色谱条件:色谱柱为Thermo BDS HYPERSIL C18 柱(250mm×4.6mm,5μm);流动相为乙腈-0.1%三氯乙酸溶液-三乙胺(18:82:0.2,磷酸调pH 至2.5);流速为1.0mL/min;检测波长为349nm,除杂质Ⅱ为317nm;柱温为25℃;进样量为20μL;灵敏度试验溶液中哌喹峰高信噪比应>10,系统适用性溶液中,理论塔板数按哌喹峰计算不低于1500,哌喹峰与杂质Ⅰ峰分离度应>14。
系统适用性溶液:取哌喹与杂质Ⅰ对照品适量,精密称定,加流动相溶解并稀释制成每1mL 中分别含1.0mg 与0.005mg 的溶液,作为系统适用性溶液。
2.2 测定方法 取青蒿素哌喹片细粉适量,加流动相溶解并稀释制成每1mL 中约含哌喹1mg 的溶液,滤过,取续滤液作为供试品溶液;精密量取1mL,置100mL 量瓶中,用流动相稀释至刻度,摇匀,作为对照溶液;精密量取对照溶液1mL,置20mL 量瓶中,用流动相稀释至刻度,摇匀,作为灵敏度溶液。取杂质Ⅰ、杂质Ⅱ与杂质Ⅲ对照品各约10mg,精密称定,分别至20mL 量瓶中,加溶剂(杂质Ⅰ加流动相、杂质Ⅱ与杂质Ⅲ加甲醇)溶解并稀释至刻度,摇匀,作为杂质Ⅰ、Ⅱ、Ⅲ对照品贮备液。分别精密量取杂质Ⅰ、Ⅱ、Ⅲ对照品贮备液各1mL,置同一100mL 量瓶中,用流动相稀释至刻度,摇匀,作为对照品溶液。精密量取上述溶液各20μL,分别注入液相色谱仪,按上述色谱条件测定,记录色谱图至主成分峰保留时间的4 倍。
2.3 专属性试验
2.3.1 专属性试验 按处方比例配制供试品溶液、哌喹及不含哌喹的阴性对照溶液,按“2.1”项下色谱条件测定,各杂质与主峰、杂质与杂质间分离度均达到要求,阴性不干扰测定,色谱图见图1-2。
2.3.2 强制降解试验 取阴性(空白辅料+青蒿素)、哌喹原料、青蒿素哌喹片细粉适量(约相当于哌喹50mg)置50mL 棕色量瓶中,加入空白溶剂25mL 溶解,加入1mol/L HCL 溶液5mL,室温放置24h 后,加入1mol/L NaOH 溶液5mL,用空白溶剂稀释至刻度,摇匀,滤过,续滤液作为酸破坏样品。取阴性(空白辅料+青蒿素)、哌喹原料、青蒿素哌喹片细粉适量(约相当于哌喹50mg)置50mL 棕色量瓶中,加入空白溶剂25mL 溶解,加入1mol/L NaOH 溶液5mL,室温放置24h 后,加入1mol/L HCL 溶液5mL,用空白溶剂稀释至刻度,摇匀,滤过,续滤液作为碱破坏样品。取阴性(空白辅料+青蒿素)、哌喹原料、青蒿素哌喹片细粉适量(约相当于哌喹50mg)置50mL 棕色量瓶中,加入空白溶剂25mL 溶解,加30%过氧化氢1mL,放置1h 后加空白溶剂稀释至刻度,摇匀,滤过,续滤液作为氧化破坏样品。各样品置105℃干燥8h后,取阴性(空白辅料+青蒿素)、哌喹原料、青蒿素哌喹片细粉适量(约相当于哌喹50mg)置50mL 棕色量瓶中,加入空白溶剂稀释至刻度,摇匀,滤过,续滤液作为高温破坏样品。取阴性(空白辅料+哌喹)、哌喹原料、青蒿素原料、按处方比例配制的供试品适量(约相当于哌喹50mg)置50mL 透明量瓶中,加入空白溶剂25mL 溶解,于5500Lx 照度的光照箱中放置24h,取出用空白溶剂稀释至刻度,摇匀,滤过,续滤液作为光照破坏样品。
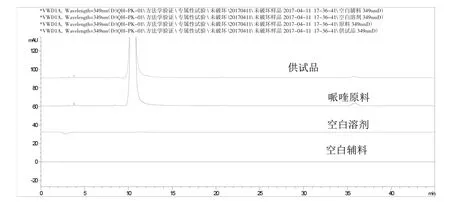
图1 专属性试验色谱图(349nm)

图2 专属性试验色谱图(317nm)
按“2.1”项下色谱条件测定,各强制降解条件下空白溶液和空白辅料对本品制剂和原料药有关物质的检测均无干扰;各强制降解条件下原料药供试品溶液中主峰与各杂质峰、各主要杂质峰之间均能有效分离;各强制降解条件下制剂供试品溶液中主峰与各杂质峰、各主要杂质峰之间均能有效分离;酸、碱、氧化、高温及强光降解条件下,哌喹原料供试品溶液的物料平衡指数分别为101.7%、101.6%、105.7%、100.8%和100.2%,制剂供试品溶液的物料平衡指数分别为98.9%、101.4%、104.0%、99.9%和100.8%;表明该方法对各主要降解杂质均能较好检出。该法能有效地检出各破坏试验产生的杂质,且各杂质峰均能与主峰达到基线分离,专属性较好。
2.3.3 滤膜吸附 方法同“2.3.1”项下专属性试验。取未过滤的供试品溶液分别滤去1、2、3、5mL,取各过滤体积下的续滤液作为滤膜吸附性供试品溶液。另取未过滤的供试品溶液适量,离心后取上清液,作为滤过供试品溶液对照。结果表明,滤膜吸附性试验中,供试品离心前后的主峰及各杂质RSD%分别为主峰:0.5%,杂质Ⅰ:0.9%,杂质Ⅱ:2.7%,杂质Ⅲ:0.5%,未知杂质1:3.7%,均不>5.0%。杂质滤过后色谱峰面积回收率均在90%~110%之间,主峰回收率>99%,说明滤膜吸附性影响较小,符合验证要求。
2.4 检测限与定量限 分别精密量取哌喹杂质Ⅰ,Ⅱ,Ⅲ对照品溶液适量,用空白溶剂逐步稀释至各样品主峰信噪比为3:1 和10:1,按“2.1”项下色谱条件测定,杂质Ⅰ的定量限和检出限分别为0.01、0.002μg/mL,杂质Ⅱ的定量限和检出限分别为0.02、0.004μg/mL,杂质Ⅲ的定量限和检出限分别为0.06、0.021μg/mL。哌喹杂质Ⅰ、Ⅱ、Ⅲ的最低检测限浓度均低于该杂质的报告限度浓度,反映该分析方法的灵敏度符合要求。
2.5 线性关系及范围 以杂质限度为100%,从定量限开始至杂质限度的400%,分别取6 个点,以浓度为横坐标(X),峰面积为纵坐标(Y),进行线性回归分析。分别精密称量哌喹杂质Ⅰ、Ⅱ、Ⅲ对照品适量,用空白溶剂溶解并稀释至每1mL 含杂质Ⅰ、Ⅱ、Ⅲ约0.2g/L 的贮备溶液,逐步稀释至浓度依次为定量限浓度(杂质Ⅰ0.01,杂质Ⅱ0.02,杂质Ⅲ0.06),0.5、1.0、2.0、5.0、8.0μg/mL,按“2.1”项下色谱条件测定,回归方程:杂质Ⅰ:y=75.9099x+0.1914,R2=1.0000;杂质Ⅱ:y=65.2220x+3.4801,R2=0.9999;杂质Ⅲ:y=95.8359x-0.2231,R2=1.0000。各线性方程的相关系数(R2)均>0.9990,Y 轴截距与100%浓度响应值之比的绝对值为0.1%,<2.0%。结果表明杂质Ⅰ浓度在0.0102~8.1800μg/mL 范围内线性关系良好,杂质Ⅱ浓度在0.0205~8.1800μg/mL 范围内线性关系良好,杂质Ⅲ浓度在0.0597~7.9600μg/mL 范围内线性关系良好。
2.6 加样回收率试验 分别精密称取青蒿素哌喹片细粉适量(约相当于哌喹100mg),置100mL 棕色量瓶中,加入各杂质贮备液,用空白溶剂溶解并稀释至刻度,摇匀,滤过,即得供试品溶液,浓度分别为供试品总量限度的50%、100%和150%,每个浓度平行制备三份平行样,按“2.1”项下色谱条件、“2.2”项下对照品溶液测定,计算加样回收率。杂质Ⅰ的加样回收率(n=9)为95.3%~96.9%,RSD 为2.0%;杂质Ⅱ的加样回收率(n=9)为90.8%~100.4%,RSD 为4.8%;杂质Ⅲ的加样回收率(n=9)为93.4%~98.7%,RSD 为3.6%。各浓度平均回收率均在90%~110%间,相对标准偏差在10%内,符合验证要求。
2.7 精密度试验 按“2.1”项下色谱条件、“2.2”项下对照品溶液测定,重复进样6 次,计算哌喹杂质Ⅰ、Ⅱ、Ⅲ峰面积的RSD 均小于0.02%,符合精密度实验要求。
2.8 重复性试验 分别精密称取青蒿素哌喹片细粉适量(约相当于哌喹50mg),置50mL 棕色量瓶中,用空白溶剂溶解并稀释至刻度,摇匀,滤过,取续滤液作为供试品溶液;平行制备6 份。按“2.1”项下色谱条件、“2.2”项下对照品溶液测定,计算哌喹杂质Ⅰ、Ⅱ、Ⅲ含量的RSD 均<0.02%,符合重复性实验要求。
2.9 溶液稳定性试验 取“2.8”项下供试品和对照品溶液,按“2.1”项下色谱条件,分别于0、4、8、16 和24h 测定,记录色谱图。24h 内供试品溶液中杂质Ⅰ、Ⅲ,未知杂质1、2 峰面积的RSD 分别为3.8%、5.7%、3.7%和0.4%,均<10%;24h 内对照品溶液中杂质Ⅰ,Ⅱ,Ⅲ峰面积的RSD 分别为0.7%、0.3%和0.7%,均<10%。各峰面积均未出现显著性变化,结果符合验证要求,表明溶液在24h 内测定结果稳定。
2.10 耐用性试验 分别考察了不同色谱柱[Thermo BDSnHYPERSILnC18(2)柱、YMC-Triart C18 柱]、不同流动相比例(乙腈-0.1%三氯乙酸-三乙胺=16:84:0.2,18:82:0.2,20:80:0.2)、不同流速(0.9,1.0,1.1mL/min)、不同三乙胺用量(0.1%、0.2%和0.3%)、不同柱温(20、25、30℃)下各色谱峰间的分离度,结果表明,柱温、流动相比例、pH 值会影响色谱峰,在样品检测时注意控制以上条件。
2.11 三批样品测定 取三批中试样品(20141001,20141002,20141003),按“2.2”项下方法制备供试品溶液与对照溶液,按“2.1”项下色谱条件测定,结果见表1。
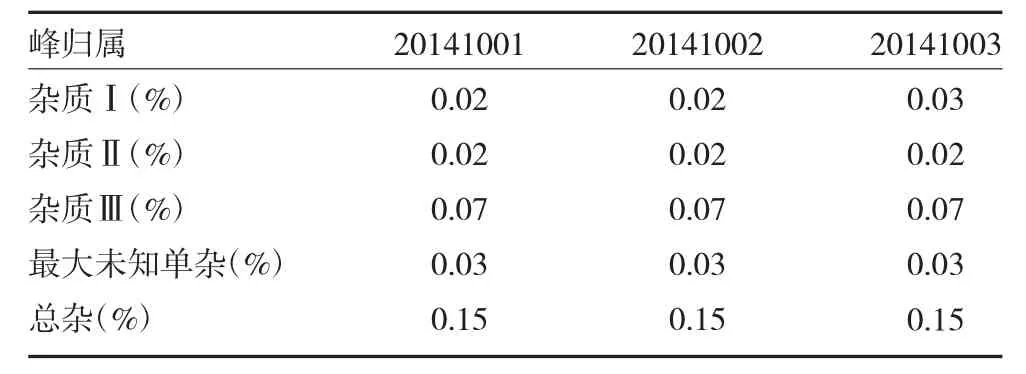
表1 三批样品有关物质测定结果
3 讨论
原方法采用自身对照法检测,均按未知杂质计算,检出结果比实际要高,且信噪比均<10,检测灵敏度低,0.05%自身对照溶液几乎不能检出,检测结果受外界影响大,可靠性不高。检测波长(216nm)处于低波段,受到外界干扰较大。
新修订方法采用原注册申报方法的色谱条件(波长除外),参考中国药典磷酸哌喹原料检测波长,349nm 检测未知杂质、杂质Ⅰ和杂质Ⅲ,317nm 检测杂质Ⅱ,采用外标法检测杂质Ⅰ,Ⅱ,Ⅲ,结果准确度高。新修订质量标准增加了药典中三个已知杂质的控制,限度均按照原料标准执行,不得过0.5%;未知杂质参考ICH 及杂质指导原则要求,不得过0.2%;总杂不得过2.0%,参考中国药典磷酸哌喹原料标准执行。依据方法验证结果及方法对比结果,参考《中国药典》2015 年版二部磷酸哌喹方法及化学药物杂质研究指导原则,制定已知杂质Ⅰ,Ⅱ,Ⅲ限度为0.5%;未知杂质由原来1.0%限度收紧,制定为0.2%;总杂质由原来2.5%收紧,制定为2.0%。
耐用性试验显示,柱温微小变化(20~25℃)对同一批次有关物质的测定结果影响不大,但达到30℃时杂质Ⅱ与主峰分离度达不到要求;色谱柱选用要求较高,方法筛选中安捷伦ZORBAX-C18 柱主峰拖尾严重,推荐使用BDS HYPERSIL C18 或类似地适用于分离碱性含氮化合物色谱柱;流动相比例的微小变化会导致主峰保留时间延后、拖尾因子变大,影响未知杂质检出,三乙胺用量需要严格定量,多或少都会影响未知杂质检出。pH 值微小变化(pH 2.5~2.7)对同一批次有关物质的测定结果影响不大,但pH 为2.3 时,主峰与杂质Ⅱ分离度达不到要求。建议使用该方法应注意流动相三乙胺用量、pH 值的控制要精准,以及要求使用固定品牌的色谱柱。
