SLC26A3基因纯合插入突变致先天性失氯性腹泻1例
2019-10-22王中华王芳刘雪芹刘爽
王中华,王芳,刘雪芹,刘爽
患儿,男,2岁9个月。因“反复腹泻、低血钾、代谢性碱中毒2年余”入院。患儿2月龄时无诱因出现大便4~10次/d,为黄色稀糊至稀水样便,无黏液、脓血及腥臭。伴呕吐、纳差,无发热。外院多次检查示低钾血症(最低2.3 mmol/L)、低氯血症(最低75 mmol/L)、代谢性碱中毒(pH最高7.66),予对症补液、口服氯化钾及特殊奶粉喂养后,患儿未再呕吐,血电解质紊乱改善,但腹泻无好转。既往史:生后因“早产、羊水过多、羊水Ⅱ°污染”住院治疗,出院诊断为“早产儿、新生儿病理性黄疸、不全性肠梗阻、先天性心脏病(室间隔缺损2 mm)”。2岁行左侧腹股沟疝修补术。个人史:第5胎第4产,胎龄35+3周,出生体质量3.15 kg,身长51 cm,生后混合喂养,腹泻后予氨基酸奶粉、“腹泻奶粉”治疗,6月龄起添加米汤、粥,并停用奶粉,目前为普食。家族史无特殊,父母非近亲结婚。入院查体:体质量12.5 kg(第10~25百分位),身长88 cm(第3百分位),其余查体无异常。辅助检查:(1)血常规正常。(2)血生化:肝、肾功能和电解质无异常,HCO3-24.2 mmol/L。(3)血pH 7.41,PaCO240.4 mmHg。(4)便常规和隐血正常,便电解质钾27.39 mmol/L,钠103.5 mmoL/L,氯138.6 mmol/L。(5)尿常规正常。尿电解质钾48 mmol/L,钠85 mmoL/L,氯39 mmol/L。(6)立位腹部X线平片示腹部多发肠管积气伴气液平(图1)。因临床考虑先天性失氯性腹泻,经患儿父母知情同意后送检SLC26A3基因分析,结果示患儿SLC26A3(NM_000111.2)基因第3外显子检测c.270_271insAA纯合、pGly91Lysfs*3(图2a),为已报道突变[1];其父母分别检测到相同杂合突变(图2b和2c)。确诊为SLC26A3基因纯合插入突变致先天性失氯性腹泻。予患儿蒙脱石散3 g/次,每日2次,双歧杆菌三联活菌散0.5g/次,每日3次口服,氯化钠(4 mmol·kg-1·d-1)及氯化钾(3 mmol·kg-1·d-1)口服治疗后大便每日4~5次(入院时7~14次),为稀水便,血电解质正常,出院后继续补充氯化钠及氯化钾。目前患儿4岁5个月,可正常上幼儿园,体质量15.5 kg(第10百分位),身高100 cm(第3百分位),每日腹泻2~4次,血电解质维持在正常范围。
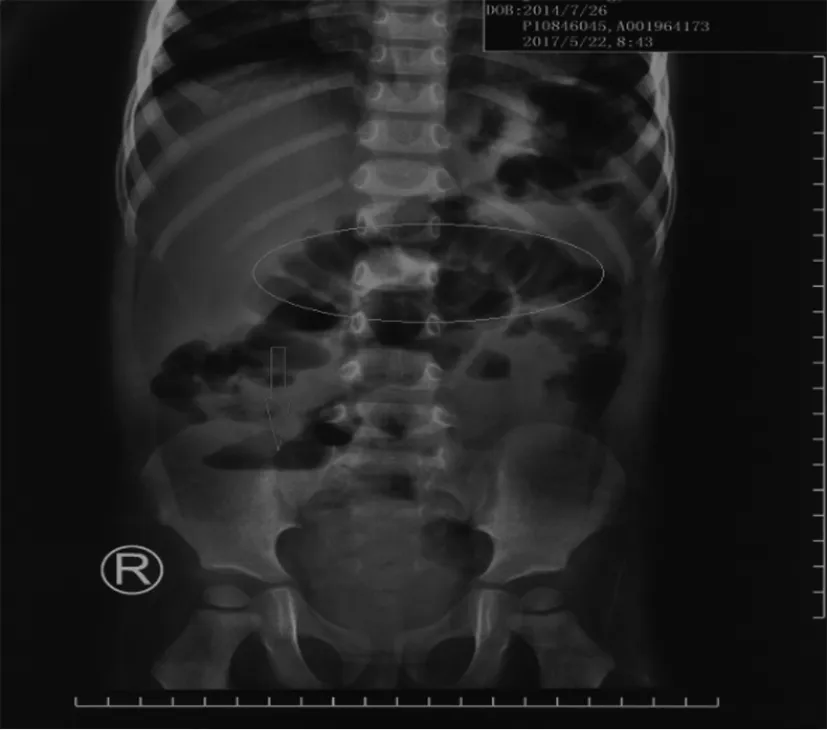
图1 患儿立位腹部X线平片示腹部多发肠管积气伴气液平
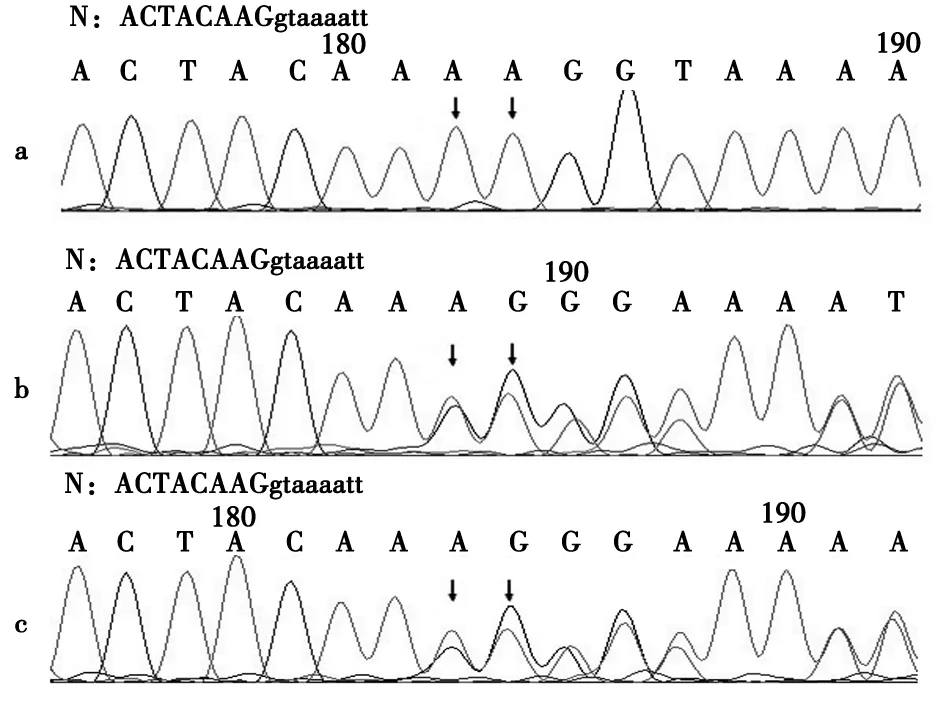
注:N.正常序列(大写字母为外显子序列,小写字母为内含子序列);↓.插入的碱基;a.患儿;b.患儿母亲;c.患儿父亲
图2 SLC26A3基因外显子3检测结果
讨 论导致婴儿及儿童慢性腹泻病的原因多种多样,包括常见的功能性腹泻、感染性疾病和少见的累及免疫调节或肠上皮细胞功能的遗传性疾病。其中基因突变致肠上皮细胞功能障碍的一类疾病通常表现为生后数月出现的大量水样便,诊治不及时将导致喂养不耐受和营养不良。因此临床医生有必要提高对此类疾病的认识。
先天性失氯性腹泻(congenital chloride diarrhea,CCD)是一种常染色体隐性遗传病,致病基因为SLC26A3基因,主要临床表现为胎儿羊水过多和肠管扩张、早产,出生后出现腹泻、低钾血症、低氯血症、低钠血症及代谢性碱中毒,大便中氯离子>90 mmol/L可以明确诊断。本例患儿胎儿期羊水多,早产,生后2个月出现水样泻,低氯血症、低钾血症和代谢性碱中毒,入院查大便中氯离子明显升高,且氯离子大于钠离子与钾离子之和,因此临床诊断CCD成立。SLC26A3基因检测分析显示本例患儿为c.270_271insAA纯合突变,父母分别携带相同杂合突变,且该突变已报道[1],故SLC26A3基因突变致CCD诊断明确。
目前在已报道的89个SLC26A3基因突变中,以错义突变、无义突变及小的缺失突变多见。国外研究显示,该病基因型和临床表型缺乏相关性[2]。截至目前国内报道的CCD病例7例,其中基因诊断5例[1,3-6],其中4例为早产儿,且3例有羊水过多病史;1例为足月儿。腹泻发生年龄为生后至2个月,2例大便氯离子>90 mmol/L。3例为纯合错义突变,2例为复合杂合突变(1例为插入突变和错义突变,另1例为无义突变和错义突变)。本例患儿临床表现典型,基因型为纯合插入突变。在有限的国人CCD患儿中未观察到基因型与表型间的相关性。值得注意的是,本例患儿尚表现有室间隔缺损和腹股沟疝,未见于国内报道的病例中。此外,有研究显示[7],具有相同基因型的同一家系不同成员临床表型不同,具有相同基因型的患者对腹泻调节剂的疗效不同。
CCD目前尚无特效治疗,获得早期诊断及补充氯化钠和氯化钾的患儿,其生长发育基本正常,预后较好。由于无法纠正原发病,腹泻可持续终生,但随年龄增长,腹泻次数减少。未经有效治疗的CCD患者可发生终末期肾脏病、肾脏钙化、高尿酸血症、腹股沟疝、精液囊肿、肠炎和男性不育[2]。因此对本例患儿进行长期管理和随访至关重要。
总之,CCD罕见,起病早,在临床实践中如果能够尽早识别并诊断该病,长期维持水电解质酸碱平衡稳定,则有助于改善其预后。
