多种遗传学技术联合运用对疑似Meckel综合征家系进行遗传学分析
2019-10-21张靓璠严恺王英黄莉
张靓璠,严恺,王英,黄莉
(1.义乌市妇幼保健院 计划生育服务中心,浙江 金华 322000;2.浙江大学医学院附属妇产科医院生殖遗传科,浙江 杭州 310000)
Meckel综合征是一种致死性的常染色体隐性遗传病,由Meckel等在1822年首次报道。典型临床症状包括肾囊肿、内生殖器异常、轴后多指畸形、肝纤维化、无脑畸形、唇腭裂、枕叶脑膨出等。
该病在不同国家新生儿中的发病率不同,在英国约为1/140 000[1],在芬兰约为1/9 000[2],在印度古吉拉特邦中发病率则高达1/1 304[3],而在我国目前仅见散发病例报道,尚无相关数据统计。该病预后差,胎儿常发生宫内死亡,出生的胎儿大多仅能存活数天至数周且没有治疗措施,因此临床上与该疾病相关的研究较少。随着基因测序技术的发展,基因检测技术已成为一种发现新致病位点和进行遗传病诊断的有效途径,并在不断地发展与完善。本研究中,我们对义乌市妇幼保健院收集到的1例疑似Meckel综合征的引产胎儿组织及其家系成员进行了遗传学检测,利用全外显子高通量测序技术及Sanger测序技术发现了CC2D2A基因上的2个致病性突变c.3964C>T和c.4567T>C。本研究丰富了Meckel综合征的致病性突变数据库,并为该家系遗传咨询、产前诊断及胚胎植入前诊断提供了遗传学依据。
1 对象和方法
1.1 对象先证者母亲,G5P2,既往体健,2007年行甲状腺结节切除术,术后恢复尚可,无高血压、糖尿病、心脏病等疾病史,无肝炎、结核等传染病史,无输血史,无明显药物、食物过敏史,无长期药物使用史,无药物成瘾。第1胎足月自然分娩女婴,健康;第2胎自愿人工流产;第3、第4胎,均为男胎,分别于妊娠13+和19+周时,B超显示枕叶脑膨出和多囊肾,经慎重考虑终止妊娠;第5胎,足月自然分娩一健康女婴。家系图谱见图1。
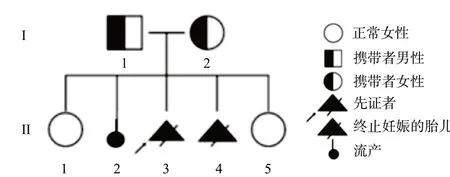
图1 Meckel综合征家系图谱
1.2 方法根据先证者胎儿超声结构异常的相关结果,我们对第3胎引产胎儿组织及其父母、姐妹进行了遗传学检测。该研究项目通过了本院医学伦理委员会的审核批准,并取得了先证者父母签署的知情同意书。取先证者胎儿腓肠肌组织,用0.9%氯化钠溶液漂洗排除母血污染,使用基因组杂交芯片(comparative genomic hybridization,CGH)和全外显子组测序(whole exome sequencing,WES)技术对先证者染色体组及外显子组进行检测;分别取先证者父母及其姐妹的EDTA抗凝外周血5 mL,针对先证者发现的致病位点,通过Sanger测序对家系进行验证。
1.2.1 CGH检测:按照Qiagen试剂盒标准流程提取血液中基因组DNA。染色体芯片采用Agilent Sure-Print G3 Custom CGH+SNP Microarray(4×180 K),操作步骤简述如下:用限制性内切酶酶切对照及样本基因组,以Cy3-dUTP和Cy5-dUTP分别标记对照和样本基因组DNA,纯化后测定标记效率及DNA量,将已标记的对照及样本基因组DNA混合,位于芯片上杂交,洗涤芯片后选择合适的扫描通道在Agilent扫描仪上扫描,扫描结果经计算机转换,得到每个位点的相对信号强度,进行作图与分析。
1.2.2 WES检测:超声将胎儿基因组DNA打断,获得200~250 bp大小的DNA片段,纯化后,对DNA片段进行末端修复、添加单碱基A以及测序接头,筛选出片段适中的DNA片段构成原始文库,PCR扩增后完成DNA建库。使用SeqCap EZ Human Exome Probes v3试剂盒,将文库与探针库进行杂交,捕获基因的外显子,洗涤后将外显子DNA富集,再次PCR扩增,纯化后得到测序文库。最后利用高通量测序仪HiseqX TEH连续双向测序(平均测序深度不低于100×),用FASTX-Toolkit软件读出原始测序数据。
1.2.3 序列分析:首先对原始测序数据进行质量评估,去除低质量及被污染的测序数据,随后用Burrows Wheeler Aligner(BWA)软件进行序列比对(比对参照hg19),同时评价序列捕获效果,用GATK软件和VarScan软件分别进行单核苷酸变异(SNP)和插入缺失(InDel)查询,得到目标区域碱基多态性结果,发现可疑的变异位点。在可疑的变异位点中去除内含子区变异,并在EXAC数据库(http://exac.broadinstitute.org)、1 000 human genome数据库(http://www.internationalgenome.org/data)中过滤频率大于1%的变异位点,添加HGMD数据库(http://www.hgmd.cf.ac.uk/ac)、OMIM数据库(http://www.omim.org)的相关注释信息,最后应用SIFT(http://sift.jcvi.org)、PolyPhen-2(http://genetics.bwh.harvard.edu/pph2)、Mutation Taster(http://www.mutationtaster.org)软件对基因突变位点的致病性进行分析,使用ExPASy(https://www.expasy.org)软件预测突变位点对蛋白结构和功能的影响,以美国医学遗传学与基因组学会(American College of Medical Genetics and Genomics,ACMG)发布的《ACMG遗传变异分类标准与指南》(2015)对发现的变异位点进行分类。
1.2.4 Sanger测序验证:采用ABI 3730测序仪验证高通量测序发现致病性突变,根据突变所在位置,在其上、下游设计相应的特异性引物。通过电泳确认PCR反应后,对片段进行Sanger测序,测序结果用Chromas软件读取,并与NCBI(https://www.ncbi.nlm.nih.gov)中的基因标准序列进行比对分析,同时验证高通量测序结果与Sanger测序结果一致性。
2 结果
2.1 CGH检测本研究首先利用CGH技术检测先证者胎儿是否存在致病性拷贝数变异、杂合性缺失或单亲二倍体。结果显示先证者胎儿染色体不存在上述3种致病性染色体异常,见图2。
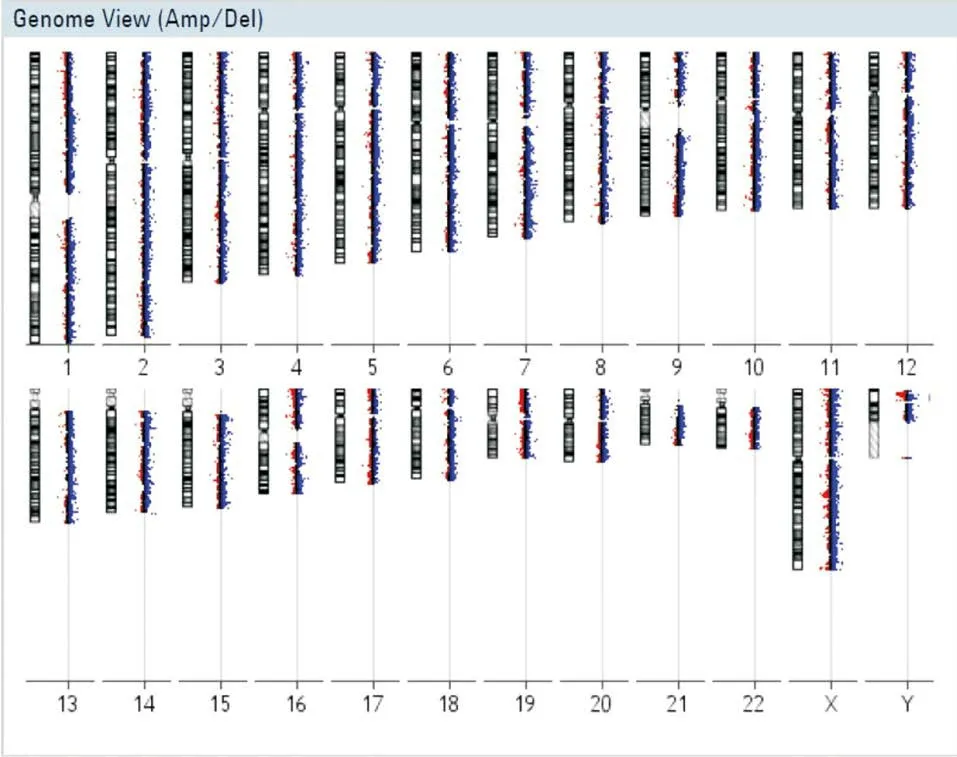
图2 先证者染色体微阵列芯片检测结果
2.2 WES检测采用全外显子测序对先证者胎儿组织的基因组DNA进行检测。检测结果显示,先证者存在CC2D2A(NM_001080522):c.3964C>T(p.Q1322X)和c.4567T>C(p.C1523R)复合杂合突变,见表1。
2.3 突变位点的功能预测c.3964C>T(p.Q1322X)为无义突变,该突变可将编码CC2D2A蛋白中第1 322号位置谷氨酰胺的密码子(CAG)变为终止密码子(TAG),从而导致编码蛋白提前终止,产生截短蛋白或蛋白被降解;该突变在人群中发生频率极低(ExAC、HGMD、ClinVar数据库均未见收录);Polyphen、SIFT、MutationTaster软件预测可能影响蛋白功能尚未见此变异在患者中检出的文献报道。根据《ACMG遗传变异分类标准与指南》(2015)将其评级为致病(PVS1+2*PP)。
c.4567T>C(p.C1523R)的变异描述和上面相似,SIFT、PolyPhen-2、Mutation Taster软件对该变异的致病性进行分析,结果均提示该突变有害;ExAC、HGMD数据库未见收录。根据《ACMG遗传变异分类标准与指南》(2015)将其评级为可能致病(2*PM+2*PP)。
2.4 Sanger 测序验证在发现先证者中存在CC2D2A基因(NM_001080522)的c.3964C>T(p.Q1322X)和c.4567T>C(p.C1523R)突变后,在先证者的父母及其姐妹中检测是否存在上述突变。Sanger测序结果显示,先证者的母亲携带CC2D2A基因c.3964C>T(p.Q1322X)突变的杂合变异。先证者父亲携带c.4567T>C(p.C1523R)杂合变异。见图3。因此先证者所携突变分别遗传自父母双方。

表1 先证者CC2D2A基因变异分析结果
3 讨论
Meckel综合征为一种罕见的致死性常染色体隐性遗传病,于1969年Opitz和Howe首次描述了Meckel综合征的临床特征[4]。该综合征的表型包括囊性肾发育不良、枕部脑膨出或其他中枢神经系统异常以及多指(趾)畸形,此3 种表型也是确诊Meckel综合征的主要标准,临床上,孕11~14周常规超声检查发现2种及以上典型的Meckel综合征表型时,即可明确诊断疾病。本研究中,先证者胎儿表现出了脑膨出和多囊肾异常,符合Meckel综合征的临床诊断标准,可诊断为Meckel综合征。
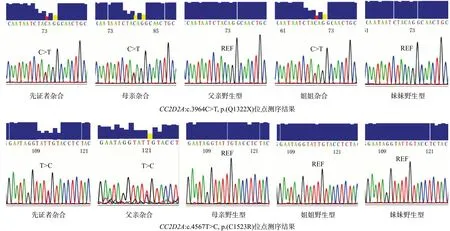
图3 家系成员CC2D2A 基因Sanger测序结果
此外,Meckel综合征与一些综合征的临床表型相互重叠,如13-三体综合征、Bardet-Biedl综合征等,临床上需要进行鉴别。13-三体综合征会出现心血管畸形及眼部异常,而很少出现肝纤维化;Bardet-Biedl综合征中不会出现脑膨出的症状[5];Joubert综合征中的典型症状是视网膜发育不良及舌组织肿瘤[6]。这些综合征除了临床表现与Meckel综合征相互重叠以外,在患者基因组中也发现了类似Meckel综合征的基因突变,因此从遗传学角度讲可能属于同一类疾病。Meckel综合征具有高度的遗传异质性,目前已知可导致Meckel综合征的致病基因有7个(MKS1、TMEM216、TMEM67、CEP290、RPGRIP1L、CC2D2A、NPHP3),分别位于不同的染色体上(17q22,11q13,8q22.1,12q21.32,16q12.2,4p15.32,3q22.1),并对应1~7型Meckel综合征。因此,基因诊断也是确诊Meckel综合征的重要手段。通过影像学方法发现胎儿异常,高度怀疑为Meckel综合征的,可收集胎儿及父母双方的样本提取DNA,利用高通量测序技术分析与Meckel综合征相关的重要基因,对可疑的遗传突变采用Sanger测序技术验证,可明确遗传病因并确定家系遗传关系。2008年首次发现Meckel综合征患者中存在CC2D2A基因的突变,在已报道的Meckel综合征患者中,CC2D2A基因突变的占比约为13%。对于Meckel综合征的致病机制,研究认为与细胞纤毛功能异常有关。纤毛是一种多见于脊椎动物细胞的突起结构,分布于除白细胞外的所有细胞表面,具有高度保守性,在细胞迁移和胞内外信号转导途径上发挥重要作用。TALLILA等[7]通过免疫荧光染色发现,CC2D2A基因突变患者的成纤维细胞中缺乏纤毛,证实了CC2D2A基因在纤毛形成中的关键作用。该基因发生突变可导致诸如微管形成、囊泡运输以及信号转导等一系列过程发生障碍的风险增加。同时,我们已知,CC2D2A基因位于人类4号染色体上,长131 692 bp,包含38个外显子,编码的蛋白质中含有一个卷曲螺旋状的钙结合结构域,此结构域在物种间高度保守,参与钙信号通路。研究发现CC2D2A蛋白在中枢神经系统中会高度表达,其功能的缺失可导致神经发育异常,进而导致疾病的发生[8]。本研究发现的c.3964C>T(p.Q1322X)导致编码谷氨酰胺的密码子突变为终止密码子,使蛋白翻译提前终止,氨基酸序列不完整,从而使蛋白功能丧失,进而影响了纤毛功能及钙信号通路,因此可能导致了Meckel综合征的发病。
根据Meckel综合征的分型和全外显子高通量测序结果,我们认为,该先证者是由CC2D2A基因突变引起的6型Meckel综合征。本研究发现,该先证者胎儿携带CC2D2A基因c.3964C>T(p.Q1322X)和c.4567T>C(p.C1523R)意义不明的复合杂合变异。在先证者母亲和父亲中分别发现并验证了CC2D2A基因c.3964C>T(p.Q1322X)、c.4567T>C(p.C1523R)的杂合突变。同时,在先证者姐姐中也发现了该基因的杂合突变,此结果提示先证者中所含有的突变来源于父母,并非新发突变。CC2D2A基因c.3964C>T(p.Q1322X)突变是一种提前终止突变,该突变在EXAC数据库和1000 human genome数据库中的出现频率均为0.0 001;c.4567T>C(p.C1523R)为意义不明突变位点,该突变在EXAC数据库和1000 human genome数据库中未有收录。SIFT、PolyPhen-2、Mutation Taster软件预测该位点突变均为有害。因此,我们认为CC2D2A基因c.3964C>T(p.Q1322X)和c.4567T>C(p.C1523R)意义不明复合杂合变异是该家系先证者患病的遗传因素。此外,我们在文献和数据库检索中未见该变异的相关报道,因此,本研究在临床上首次发现了CC2D2A 基因c.3964C>T(p.Q1322X)和c.4567T>C(p.C1523R)意义不明复合杂合突变与Meckel综合征的联系。通过对Meckel综合征一个相关家系的临床及遗传学分析明确了Meckel综合征的临床和基因诊断信息,CC2D2A基因变异很可能是该Meckel综合征患病家系的致病原因,但仍需通过蛋白质组学研究进一步证实。此外,该研究重点对于已发现的遗传病致病基因进行筛查,并不能完全排除其他少见或目前未发现的致病基因导致Meckel综合征的可能性。我们的发现对于Meckel综合征的确诊及家系遗传咨询,进一步探索其发病机制、表型-基因型相关性,发现治疗靶点及指导临床工作具有重要意义。
生育过Meckel综合征患儿的夫妇再次生育时的再发风险为25%。因此,及时发现并明确先证者患儿致病原因可为夫妇再次生育提供指导,减少出生缺陷儿的出生。胚胎植入前遗传学诊断(preimplantation genetic diagnosis,PGD)是遗传性出生缺陷的源头阻断技术。本研究的家系中先证者的致病变异及来源明确,据此可采用PGD技术阻断该致病变异在家系中的世代遗传。
临床上,患有遗传综合征的胎儿通常表现出超声结构异常,由于存在遗传异质性,相同的临床表型可能由不同的致病基因突变所致。综合其他研究及本研究的结果,基于二代测序的WES检测已成为对此类胎儿进行遗传学检测的重要策略。
