GPC3-WNT-JNK通路介导脂多糖诱导肺局部细胞炎症
2019-10-21陈超蕾林雨婷张桑桑宋晨剑何飞董莉陈俊杰王蓓蓓董年李玉苹陈成水
陈超蕾,林雨婷,张桑桑,宋晨剑,何飞,董莉,陈俊杰,王蓓蓓,董年,李玉苹,陈成水
(1.温州医科大学附属第一医院 呼吸与危重症医学科,浙江 温州 325015;2.温州市中西医结合医院心血管内科,浙江 温州 325000)
急性肺损伤(acute lung injury,ALI)是一种常见、严重的临床疾病,其发展至严重阶段时被称为急性呼吸窘迫综合征(acute respiratory distress syndrome,ARDS)[1]。致病因素可导致肺泡-毛细血管细胞屏障的非特异性损伤,从而造成弥漫性肺间质及肺泡水肿,最终引起急性低氧性呼吸功能不全或衰竭。其发病机制错综复杂[2],涉及中性粒细胞等炎症细胞肺内浸润、肺血管内皮细胞和气管上皮细胞、肺泡上皮细胞的损伤、肺间质成分及肺泡水肿等[3]。我们的肺损伤动物模型研究表明:气道内局部给予磷脂酰肌醇-3-羟激酶(phosphatidylinositol 3-hydroxykinase,PI3K)抑制剂比全身给药更能预防和减轻脂多糖(lipopolysaccharide,LPS)及胰蛋白酶诱导的肺损伤作用,提示气道局部炎症可能是肺损伤发生发展的重要环节[4]。
近期,我们在研究临床ALI/ARDS患者血液蛋白谱检测时,发现并筛选出许多ALI/ARDS相关特异性蛋白[5]。其中磷脂酰肌醇蛋白多糖-3(glypican-3,GPC3)在肺炎相关ALI/ARDS患者血清中含量急剧升高。但GPC3在ALI中扮演何种角色,其通过何种信号途径参与肺损伤疾病的发生尚不明确。
本研究采用LPS经气道滴入制造小鼠ALI模型后研究GPC3表达水平及位置,随后予LPS刺激人肺上皮细胞16HBE,研究局部细胞炎症时GPC3及各种炎症因子的表达,同时给予外源性GPC3,研究GPC3对肺局部细胞炎症的影响,通过抑制GPC3-WNT通路,分析GPC3-WNT通路在局部炎症的调控作用,初步探讨相关的分子机制,为明确GPC3在LPS诱导的ALI中的作用及涉及的信号途径提供理论依据。
1 材料和方法
1.1 材料
1.1.1 实验动物:20~25 g(6~8周)雄性C57BL/6J小鼠由温州医科大学实验动物中心提供,实验动物许可证号:SYXK(浙)2015-0009。小鼠置于温州医科大学实验动物中心SPF级实验动物房进行饲养及实验。室内温度控制在20~25 ℃,湿度控制在40%~70%。
1.1.2 实验材料:LPS(L9143)购自美国Sigma公司;外源性GPC3(10088-H08H)购自北京Sino Biological公司;SYBR Green Real-time PCR Master Mix-Plus购自日本TAKARA公司;BCA蛋白浓度测定试剂盒购自美国Thermo公司;GPC3 ELISA试剂盒(EM1084)购自美国Fine Test公司;TNF-α ELISA试剂盒(MTA00B)购自美国Rffamp;D Systems公司;Anti-Glypican3 抗体(ab66596)、Anti-EpCAM抗体(ab71916)购自英国Abcam公司,Anti-CD31抗体(GB13063)、驴抗兔抗体(GB22403)、驴抗山羊抗体(GB21404)购自武汉Servicebio公司,山羊抗兔抗体(B0015K122300)购自合肥Biosharp公司。Anti-TNF-α抗体(AF7014)、Anti-TGF-β抗体(AF0260)购自常州Affinity公司。SP600125(S1460)购自英国Selleck Chemicals公司。Real-time PCR涉及的引物均购自上海生工公司。
1.2 方法
1.2.1 ALI小鼠模型的建立及鉴定:选择C57BL/6健康雄性小鼠,腹腔注射10%水合氯醛(4 mL/kg)麻醉,仰卧位固定于有60°夹角的滴注板上,用接注射器的软管吸除口咽部分泌物,左手用扁平镊子拉出小鼠舌头,纱布包裹固定,右手用镊子撑开小鼠咽部,即可看到声门,将带针芯的静脉留置针(隐藏锐利针头)插入气管内,立即拔出针芯,剪取小鼠毛发一根,将其置于气管插管通气口上方,观察毛发随呼吸气流上下运动,即可判断插管位于气道内。向气管内快速滴注LPS(浓度1 000 μg/mL,注射量4 mg/kg)或PBS,并立即注入空气0.5 mL。给药结束后,置于取暖器前照射,以维持小鼠体温。待小鼠完全苏醒后,放回饲养笼内,给予食物和水。4、24 h后,予以麻醉后收集小鼠气道肺泡灌洗液(bronchoalveolar lavage fluid,BALF)及静脉血,处死小鼠,取出肺组织,右肺用于RT-PCR及Western blot,左肺用于冰冻切片和石蜡切片。应用全自动血液学分析仪计数BALF中白细胞、中性粒细胞总数及总蛋白水平。HE染色鉴定急性肺损伤小鼠造模炎症表达。
1.2.2 免疫荧光观察GPC3表达:冰冻切片从冰箱取出复温,晾干水分,多聚甲醛固定60 min,待多聚甲醛完全干后于PBS(pH 7.4)中在脱色摇床上晃动洗涤3次,每次5 min。组织切片置于盛满EDTA抗原修复缓冲液(pH 8.0)的修复盒中于微波炉内进行抗原修复。低火10 min,此过程需防止缓冲液过度蒸发,切勿干片。自然冷却后将玻片置于PBS(pH 7.4)中在脱色摇床上晃动洗涤3次,每次5 min。加入自发荧光淬灭剂5 min,流水冲洗10 min。后滴加BSA孵育30 min,轻轻甩掉封闭液,在切片上滴加PBS按一定比例配好的一抗(GPC3 1:500或CD31 1:100或EpCAM 1:100),切片平放于湿盒内4 ℃孵育过夜。玻片置于PBS(pH 7.4)中在脱色摇床上晃动洗涤3次,每次5 min。切片稍甩干后在圈内滴加与一抗相应种属的二抗覆盖组织,避光室温孵育50 min。玻片置于PBS(pH 7.4)中在脱色摇床上晃动洗涤3次,每次5 min。切片稍甩干后用抗荧光淬灭封片剂封片,切片于荧光显微镜下观察并采集图像。
1.2.3 免疫组织化学观察GPC3表达:予以左肺石蜡切片、脱蜡、水化、阻断、抗原修复并进行通透。随后滤纸吸除组织切片上多余液体,滴加适量封闭液,5% BSA或2%山羊血清溶液室温放置30 min。滤纸吸除组织切片上多余液体后,滴加适量Anti-GPC3抗体(1:100)4 ℃冰箱过夜或室温放置1~2 h。组织切片浸泡PBS溶液5 min,重复3次,洗去一抗。滤纸吸除多余液体后,滴加HRP标记的山羊抗兔二抗(1:5 000),室温或37 ℃放置30 min后,浸泡PBS溶液5 min,重复3次,洗去二抗。滤纸吸除组织切片上多余液体后,滴加适量DAB显色液5~10 min,同时在显微镜下观察显色情况,根据情况及时停止反应,自来水冲洗5 min。组织切片浸入苏木素染液中染色5 min后,再滴加1%盐酸酒精浸泡分化2~3 s,使用PBS缓冲液浸泡返蓝3 min后,染色、分化、反蓝后均用自来水冲洗20 s。组织切片脱水透明,在载玻片上滴加适量中性树脂后,盖上盖玻片,显微镜下检测肺组织中GPC3表达水平。
1.2.4 细胞培养:用含10%胎牛血清的PMI-1640培养液基于37 ℃、5% CO2的恒温条件下培养16HBE细胞,并用0.25%的胰蛋白酶消化传代。
1.2.5 Real-time PCR检测不同浓度的LPS刺激16HBE细胞不同时间后GPC3及各种炎症因子的mRNA表达水平:不同浓度LPS(0.1、1、5 μg/mL)刺激人肺上皮细胞16HBE细胞不同时间(0.5、4、12、24 h)后,加入1 mL TRIzol(每106个细胞加1 mL),裂解反应10 min,加1 mL的三氯甲烷溶液,4 ℃、12 000 r/min离心15 min,吸取400 μL的上层水相层溶液与等体积的异丙醇混合后,静置10 min,4 ℃、12 000 r/min离心15 min,弃上清,用75%的乙醇溶液洗涤2次后,在室温下自然晾干,用紫外分光光度计检测提取的RNA的浓度及纯度。Realtime PCR检测GPC3、TGF-β和TNF-α表达,程序为94 ℃预变性5 min; 94 ℃变性20 s、58 ℃退火30 s、72 ℃延伸2 min,40个循环;72 ℃延伸5 min。引物序列见表1。以GAPDH为内参照,2-ΔΔCt法计算GPC3、TGF-β和TNF-α的mRNA表达水平。
1.2.6 Western blot检测不同浓度的LPS刺激16HBE细胞不同时间后GPC3及各种炎症因子蛋白表达水平:将上述各组细胞中加入蛋白裂解液,置于冰上裂解30 min后,14 000 r /min,4 ℃离心20 min,将蛋白上清转移到EP管中。用BCA法对蛋白样品进行定量,步骤参照BCA蛋白定量检测试剂盒。将蛋白样品与等量的2×Loading Buffer混合煮沸5 min。10%分离胶和5%浓缩胶进行凝胶电泳,每孔上样量为50 μL。在浓缩胶中用90 V电压电泳,在分离胶中用120 V电压电泳。4 ℃将蛋白转印至硝酸纤维素膜上。用5%脱脂奶粉室温封闭1 h,依次与I抗(1:1 000,4 ℃过夜)和II抗(1:5 000,室温1 h)孵育后,显色,以GAPDH为内参照,分析GPC3、TGF-β和TNF-α的蛋白水平。

表1 Real-time PCR引物序列
1.2.7 外源性GPC3刺激16HBE细胞后各炎症因子的表达水平:将不同浓度的GPC3(50、250、500 ng/mL)刺激16HBE细胞24 h后,分别用Real-time PCR检测TGF-β和TNF-α mRNA表达及Western blot检测蛋白表达情况,具体步骤参照1.2.5及1.2.6。
1.2.8 GPC3-WNT通路抑制剂对GPC3刺激16HBE细胞后炎症因子的影响:将不同浓度WNT/JNK通路抑制剂SP600125预处理16HBE细胞,再予以外源性GPC3刺激24 h后,分别用Real-time PCR检测TGF-β和TNF-α mRNA表达及Western blot检测TGF-β和TNF-α蛋白表达情况,具体步骤参照1.2.5及1.2.6。
1.3 统计学处理方法
采用SPSS13.0软件进行统计学分析,GraphPad Prism 6、Image Lab进行数据记录、提取、图表绘制。计量资料以±s 表示,多组比较采用单因素方差分析,组间两两比较用LSD检验。P<0.05为差异有统计学意义。
2 结果
2.1 LPS诱导小鼠产生ALI/ARDS炎症反应
为评估肺部炎症和内皮屏障功能障碍,我们检查从血液循环进入肺泡空间的白细胞数量,并以肿瘤坏死因子α(tumor necrosis factor-α,TNF-α)作为系统性炎症介质的指标。结果表明与PBS处理小鼠相比,LPS处理小鼠气管内4 h时肺间质和肺泡内炎性细胞浸润增加,随着时间的推移,LPS处理24 h的小鼠肺内白细胞迁移似乎比LPS处理4 h的小鼠更为明显,见图1a。ALI小鼠气道BALF中白细胞总数、中性粒细胞数、蛋白水平和TNF-α表达水平较PBS组显著升高(P<0.01),且随着LPS刺激时间的延长,呈上升趋势(见图1b-e)。
2.2 LPS诱导ALI/ARDS小鼠模型中GPC3 的表达
免疫组织化学染色显示,与PBS组比,LPS诱导4、24 h后ALI小鼠肺组织GPC3的免疫反应活性更显著,GPC3蛋白表达的定位基本存在于支气管上皮细胞中,见图2。LPS诱导后,ALI小鼠肺组织GPC3 mRNA表达明显高于对照组,在LPS刺激4 h时达到峰值(P<0.05),见图3a。LPS组BALF中GPC3蛋白的表达高于PBS组(P<0.05),见图3b。4 h或24 h LPS诱导的小鼠肺组织中的GPC3蛋白表达明显高于PBS组(P<0.05),见图3c。为研究哪种特定细胞参与GPC3的表达,于LPS刺激后肺组织进行免疫荧光染色,分别予以上皮细胞特定标记物EpCAM标记(红色)、内皮细胞特定标记CD31(红色)和GPC3(绿色),发现GPC3主要是在肺支气管上皮细胞、肺泡上皮细胞上表达,其中以支气管上皮细胞为主,见图4。
2.3 LPS诱导16HBE细胞中GPC3及炎症因子等表达
通过Real-time PCR检测,不同浓度(0.1、1、5 μg/mL)不同时间点(0.5、4、12、24 h)LPS刺激人肺支气管上皮细胞16HBE细胞后GPC3、TNF-α、TGF-β的mRNA表达均有不同程度的升高。GPC3的mRNA表达随着LPS浓度的增高及LPS刺激时间的延长,于1 μg/mL 24 h、5 μg/mL 4 h、5 μg/mL 12 h、5 μg/mL 24 h明显升高,与PBS组比差异有统计学意义(P<0.05),见图5a。TNF-α mRNA表达随着LPS浓度的增高及LPS刺激时间的延长,于1 μg/mL 24 h、5 μg/mL 12 h、5 μg/mL 24 h升高,与PBS组比差异有统计学意义(P<0.05),见图5b。TGF-β的mRNA表达则随着LPS浓度的增高及LPS刺激时间的延长,于1 μg/mL 12 h、1 μg/mL 24 h、5 μg/mL 12 h、5 μg/mL 24 h升高,与PBS组比差异有统计学意义(P<0.05),见图5c。
予以不同浓度LPS刺激人肺16HBE细胞24 h,应用Western blot法检测GPC3、TNF-α、TGF-β蛋白分泌水平。与对照组相比,GPC3于5 μg/mL 24 h组升高,差异有统计学意义(P<0.05)。TNF-α和TGF-β则随着LPS浓度升高,蛋白表达亦升高,其中1 μg/mL 24 h组及5 μg/mL 24 h组均较对照组明显升高,差异有统计学意义(P<0.01)。见图6。
2.4 外源性GPC3刺激16HBE细胞中炎症因子等表达
通过Real-time PCR检测,不同浓度(50、250、500 ng/mL)外源性GPC3刺激人肺支气管上皮细胞16HBE细胞24 h后,TNF-α、TGF-β的mRNA表达均有不同程度的升高。其中TNF-α mRNA表达在外源性GPC3浓度为500 ng/mL时较PBS组升高(P<0.05),见图7a。TGF-β mRNA表达随着外源性GPC3浓度的增高于250 ng/mL及500 ng/mL均有升高,与PBS组比差异有统计学意义(P<0.05),见图7b。Western blot检测结果显示,与PBS组比,500 ng/mL GPC3组TNF-α蛋白表达明显升高,差异有统计学意义(P<0.01)。TGF-β蛋白表达则随着外源性GPC3浓度升高亦升高,其中250 ng/mL及500 ng/mL组均较PBS组升高明显,差异有统计学意义(P<0.01),见图7c。2.5 GPC3通过WNT-JNK通路介导16HBE细胞炎症因子表达 予以不同浓度(20、40 mmol/L)WNT/JNK通路的抑制剂SP600125预处理16HBE细胞2 h后加入500 ng/mL GPC3刺激16HBE细胞24 h,采用Realtime PCR检测TNF-α、TGF-β的mRNA表达。TNF-α mRNA于500 ng/mL GPC3组较PBS组升高(P<0.05),而加入通路抑制剂SP600125 处理后500 ng/mL GPC3+20 mmol/L SP组和500 ng/mL GPC3+40 mmol/L SP组的TNF-α mRNA表达均较500 ng/mL GPC3组下降,差异有统计学意义(P<0.01),见图8a。TGF-β的mRNA表达变化情况与TNF-α类似,均在500 ng/mL GPC3组较PBS组升高(P<0.05),而500 ng/mL GPC3+40 mmol/L SP组较500 ng/mL GPC3组下降,差异有统计学意义(P <0.05)见图8b。通过Western blot法检测TNF-α、TGF-β蛋白表达水平可发现,SP600125预处理过的16HBE通过GPC3刺激后,TNF-α和TGF-β蛋白水平较500 ng/mL GPC3组有所下降,其中40 mmmol/L浓度的SP600125预处理后明显下降,差异有统计学意义(P<0.05),见图8c。
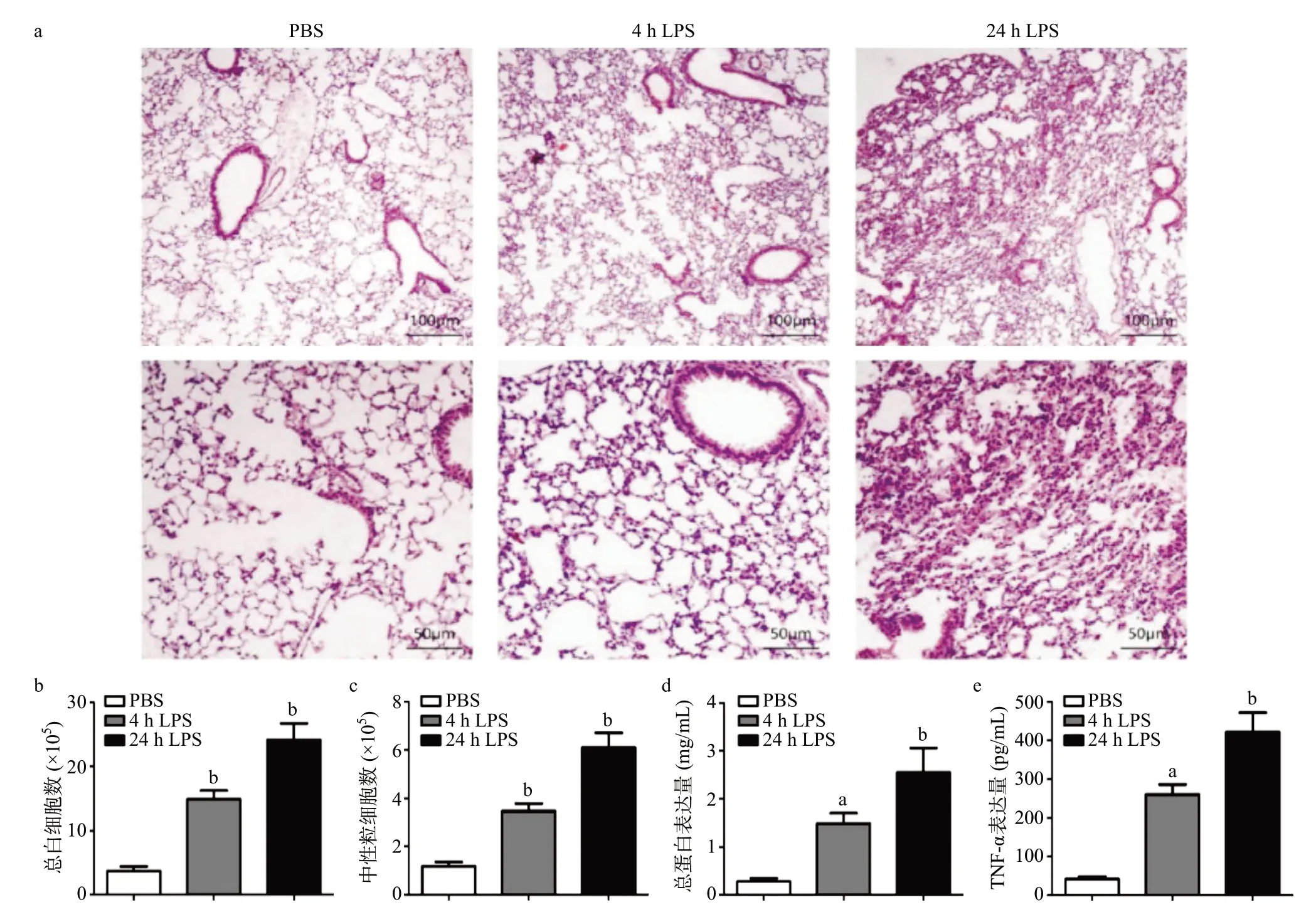
图1 LPS诱导ALI小鼠炎性细胞浸润及炎症因子产生

图2 LPS诱导ALI小鼠GPC3免疫组织化学染色结果(大图×40,右下角小图×200)

图3 LPS诱导ALI小鼠GPC3表达
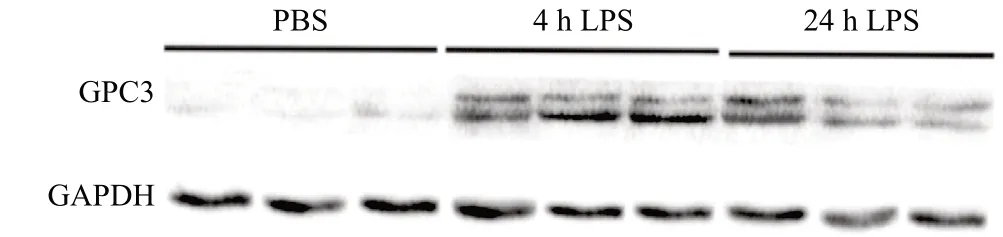
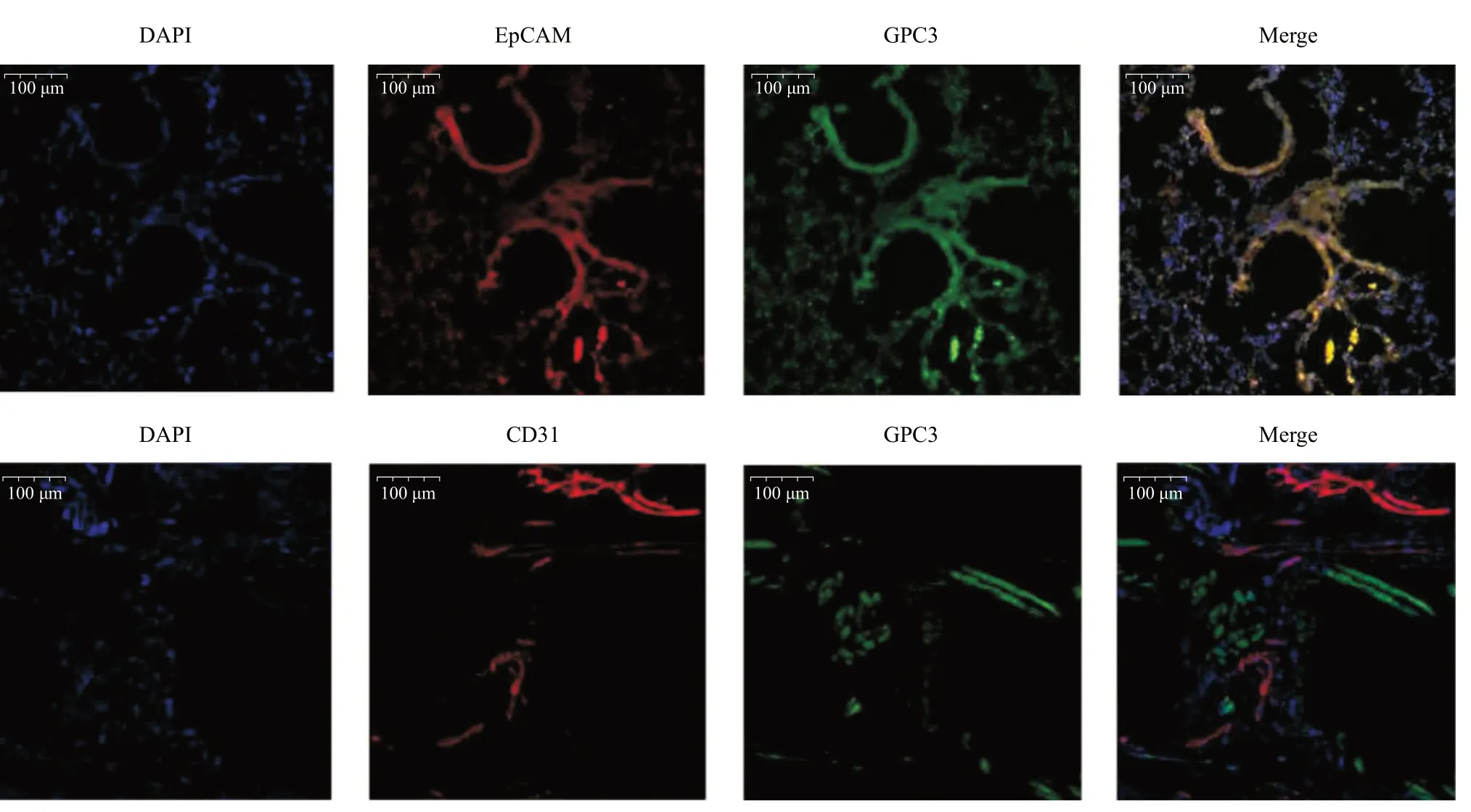
图4 LPS处理4 h后小鼠肺组织GPC3免疫荧光染色图
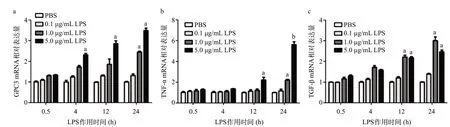
图5 不同浓度LPS刺激16HBE细胞不同时间后GPC3(a)、TNF-α(b)、TGF-β(c)mRNA表达水平
3 讨论
ALI/ARDS是临床上常见可危及生命的急性病,其特征是过度的炎症反应和肺泡上皮-毛细血管屏障的损伤,其发病过程中涉及了诸多不同的细胞和相关的炎症介质[5]。
近期,我们在研究ALI/ARDS患者血液蛋白谱检测时,发现并筛选出许多ALI/ARDS疾病相关特异性蛋白。在研究这些与ALI具有潜在相关性的蛋白时,发现GPC3在肺炎相关ALI/ARDS患者血清中含量急剧升高[5]。进一步研究GPC3在气管滴入LPS诱导肺损伤动物模型的肺内表达情况时发现,ALI时GPC3的表达升高明显,且GPC3主要定位于肺内的气道上皮细胞,亦表达于少许肺泡细胞。
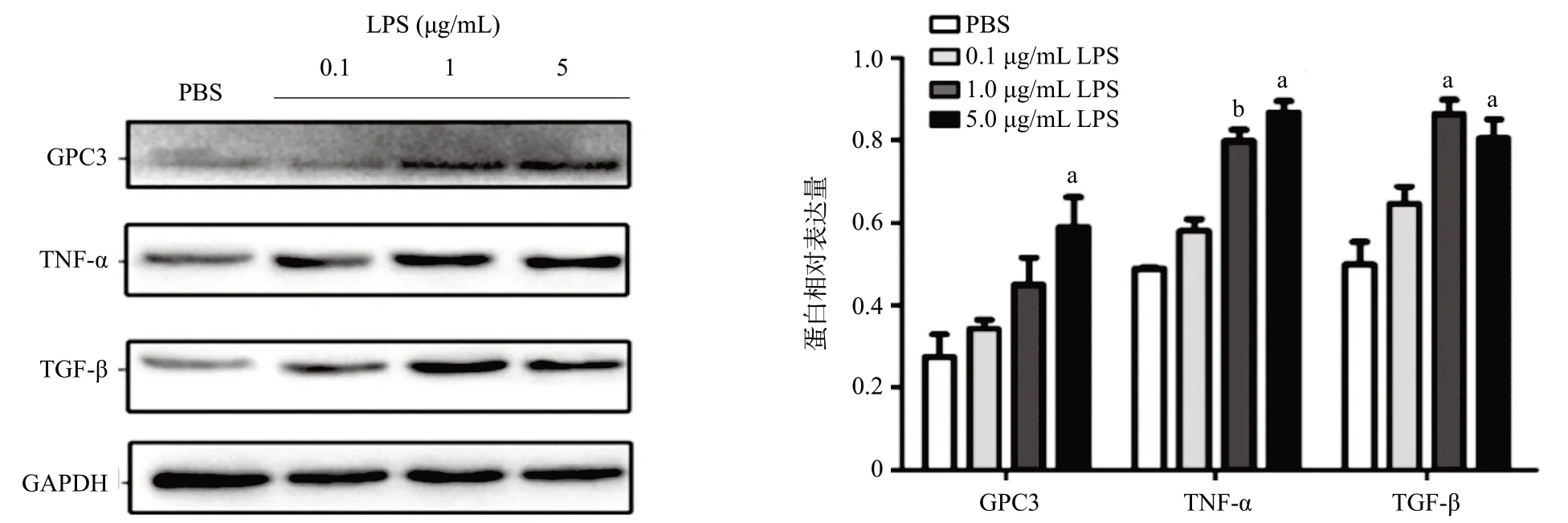
图6 不同浓度LPS刺激16HBE细胞24h后GPC3、TNF-α、TGF-β蛋白表达情况
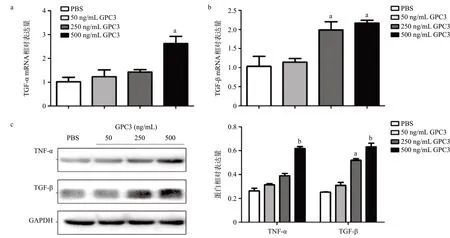
图7 不同浓度外源性GPC3刺激16HBE细胞24 h后TNF-α(a)、TGF-β(b)mRNA及TNF-α、TGF-β(c)蛋白表达
GPC3为Glypican家族的一员[6-7],为膜性硫酸乙酰肝素多糖蛋白,国内外研究均聚焦于其可通过影响多个分子信号通路调控组织器官发生、生长的重要作用上。研究表明,GPC3作为一种癌胚蛋白在多种肿瘤中过表达,如肝癌[8-9]、肺癌[10]、Wilms瘤[11]等。近期,BOSWELL等[12]研究发现GPC3衍生肽-15氨基酸长度的GPC81-95,可诱导CD4+T细胞及外周血单个核细胞产生TGF-β,具有一定的调控炎症效果。但该研究未深入探讨GPC81-95这种GPC3衍生肽调节炎症因子的具体通路及机制。
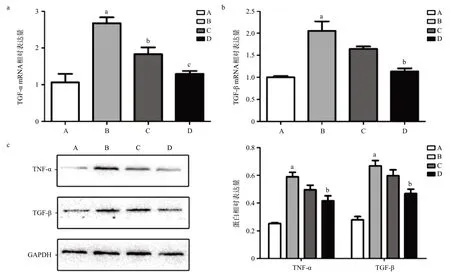
图8 不同浓度SP600125预处理16HBE细胞后GPC3刺激24 h后TNF-α、TGF-β mRNA(a-b)及蛋白(c)表达
肿瘤相关研究表明,GPC3可参与多种信号通路调节、调控恶性肿瘤的生长、增殖,如WNT/JNK信号通路[13-15]、Hedgehog分子信号通路[16-17]、Integrin信号通路[18]等。WNT是一类分泌型糖蛋白,其信号通路是一类传导生长刺激信号的通路,WNT/JNK通路是WNT涉及的一条通路,其可调控细胞骨架的重排[19],主要作用是对胚胎发育的阶段性调控。JNK又被称为应激活化蛋白激酶,是哺乳类细胞中MAPK信号通路的一种亚类[20]。既往研究证实,JNK通路可通过C-Jun和ATF-2磷酸化,调节NF-κB的激活,启动一系列与应激相关的炎性介质的合成[21]。JNK信号转导通路与细胞应激、凋亡以及纤维化密切相关,抑制JNK信号通路,可减弱ALI/ARDS中炎症介质的释放,减少中性粒细胞在急性肺损伤动物模型中肺部浸润,进而缓解炎症反应,改善生存率[22-23]。在缺血/再灌注动物模型中,JNK信号通路可介导线粒体释放的促凋亡因子,直接磷酸化Bcl促凋亡家族的成员,诱导促炎细胞因子TNF-α、IL-1的表达[24]。以上研究表明,JNK信号通路与炎症信号调控密切相关。
本研究首先通过气道滴入LPS成功模拟小鼠ALI模型,并检测ALI小鼠肺组织及BALF中GPC3表达情况,结果显示LPS刺激下的ALI过程中GPC3表达升高,同时GPC3主要表达于上皮细胞尤其支气管上皮细胞。通过LPS刺激人支气管上皮细胞16HBE检测GPC3和2种炎症相关因子的表达,结果提示一定浓度的LPS刺激16HBE细胞创建炎症微环境时GPC3有一定程度的升高,证实GPC3与LPS诱导肺局部细胞形成的炎症微环境有一定关系。同时,16HBE细胞中予以不同浓度GPC3刺激24 h后,发现TGF-β和TNF-α表达均随着GPC3浓度的升高而升高,呈一定的剂量依赖性,提示GPC3对肺内局部细胞存在着重要的促炎作用,可能是LPS刺激肺局部细胞炎症微环境中的重要一环。
为进一步探讨GPC3在LPS诱导肺局部细胞炎症微环境中可能涉及的调控通路,应用WNT/JNK通路抑制剂SP600125预处理细胞后,加入外源性GPC3蛋白刺激24 h后,发现抑制剂处理组中的各种炎症因子如TGF-β、TNF-α mRNA及蛋白表达水平均较GPC3组下降,结果提示GPC3可通过WNT-JNK通路调节肺局部细胞的炎症因子表达水平。
本研究通过小鼠ALI模型揭示GPC3在ALI中的表达情况,并定位其主要表达细胞即上皮细胞,阐述GPC3 是LPS诱导局部炎症反应中重要的环节。WNT/JNK通路抑制剂可一定程度上抑制GPC3对肺局部细胞的促炎作用,提示GPC3-WNT/JNK参与肺局部细胞的急性炎症反应。这些结果提示GPC3可能作为促炎基因参与LPS诱导的ALI炎症的发生发展,并可能成为一种新的ALI判断标志物及治疗靶点。
