Hepatic senescence, the good and the bad
2019-09-25NazmulHudaGangLiuHonghaiHongShengminYanBilonKhambuXiaoMingYin
Nazmul Huda, Gang Liu, Honghai Hong, Shengmin Yan, Bilon Khambu, Xiao-Ming Yin
Abstract Gradual alterations of cell’s physiology and functions due to age or exposure to various stresses lead to the conversion of normal cells to senescent cells. Once becoming senescent, the cell stops dividing permanently but remains metabolically active. Cellular senescence does not have a single marker but is characterized mainly by a combination of multiple markers, such as,morphological changes, expression of cell cycle inhibitors, senescence associated β-galactosidase activity, and changes in nuclear membrane. When cells in an organ become senescent, the entire organism can be affected. This may occur through the senescence-associated secretory phenotype (SASP). SASP may exert beneficial or harmful effects on the microenvironment of tissues. Research on senescence has become a very exciting field in cell biology since the link between age-related diseases, including cancer, and senescence has been established. The loss of regenerative and homeostatic capacity of the liver over the age is somehow connected to cellular senescence. The major contributors of senescence properties in the liver are hepatocytes and cholangiocytes. Senescent cells in the liver have been implicated in the etiology of chronic liver diseases including cirrhosis and hepatocellular carcinoma and in the interference of liver regeneration. This review summarizes recently reported findings in the understanding of the molecular mechanisms of senescence and its relationship with liver diseases.
Key words: Senescence; Senescence associated secretory phenotype; Hepatocyte;Cholangiocyte; Hepatic stellate cell; Cell cycle arrest; DNA damage
INTRODUCTION
Definition of senescence
In response to endogenous and exogeno us stress, cells in various tissues may enter into a permanent cell cycle arrest. These cells cannot proliferate but remain metabolically active for an extended period of time and are thus termed as senescent cells. Conversion of normal cells into senescent cells occurs throughout the life-time of an organism and plays important roles in tissue homeostasis[1]. In 1961, Leonard Hayflick observed that a human fibroblast cell ceased replication after forty to sixty divisions. As a result of Hayflick’s experiments the limited capacity for cellular division in cell culture was termed as the “Hayflick Limit”. Later it was found that the cause of the Hayflick Limit was the shortening of telomeres, portions of DNA at the ends of chromosomes, which slowly erode as cells replicate.
Hayflick and Moorhead first introduced the term senescence to describe the limited proliferation ability of human fibroblasts in cell culture. However, cells with senescent properties have been discoveredin vivoin many types of tissues and the number of senescent cells increases with age[1-6]. Initially senescence was thought to be an artifact in tissue culture with no relevance to the physiology and pathology of an organism.But subsequent studies have proved the importance of senescence in the biological processes, such as embryonic development, tissue repair, tumor suppression and aging[7]. Senescence is now considered as a multistep, dynamic cellular process. When cells are stimulated by senescence-inducing signals, such as oncogene activation,DNA damage, or other stress-mediated signals, they undergo cell cycle arrest or senescence initiation. In the next step, cells with cell cycle arrest undergo chromatin remodeling, present senescence-associated secretory phenotype (SASP), change morphology and gain other characteristics of a full-fledged senescence phenotype.Senescent cells can persist for months[8]. When senescence occurs due to oncogene activation, cell cycle arrest first happens in an autocrine manner, but SASP factors can induce paracrine senescence in other cells at a late stage after the immune system is activated[9,10], thus spreading senescence to neighboring cells across an organ.
There are two hypotheses to explain whether cellular senescence is beneficial or detrimental to an organism. We can consider senescence as a tumor suppressive or anti-cancer process as the senescent cells cannot divide. Therefore, senescence may be good for an organism. On the other hand, cellular senescence may cause loss of regenerative capability of an organ such as the liver. In this respect, it is considered a deleterious process for an organism as it may affect the function and tissue renewal[11].In either case, morein vivostudies on the mechanisms are needed.
Characteristics of senescence
The senescence phenotype is very stable, unresponsive to mitogenic stimuli and resistant to apoptosis[11]. The telomere dysfunction in normal cells can lead to the conversion to replicative senescent cells. Important stimuli like oncogene overactivation, global DNA damage, and oxidative stress may act individually or synergistically to trigger senescence in normal cells[12]. When cells undergo senescence,they are characterized by alterations in morphology, lysosomal activity and geneexpression. These include expression of cell cycle inhibitors, such as p15INK4B, p16INK4A,and p21Cip1, activation of DNA damage response, alterations of chromatin structures and induction of SASP. Usually senescent cells exhibit enlarged, flat morphology and are frequently multi-nucleated[13]. For example, the report from Aravinthanet al[14]showed that senescent hepatocytes, which overexpress p21Cip1, can be characterized with larger nuclei, compared to non-senescent hepatocytes.
Senescent cells express a higher level of lysosomal β-galactosidase gene (GLB1) and show an increased β-galactosidase activity at a suboptimal pH of 6.0 (optimal pH level is 4-4.5), compared to normal cells[3,15]. Of note, as a single cell based assay, the βgalactosidase activity assay is the most commonly used analysis for the senescence property of cellsin vitro or in vivo[16]. The cumulative effect of various stresses on the small population of affected cells may lead to conversion of a homogenous population of senescent cells[12].
SASP and senescence
Senescent cells can secrete a large number of proinflammatory cytokines, chemokines,growth factors and proteases that may have a variety of effects on neighboring cells.This phenotype of senescent cells is known as senescence-associated secretory phenotype (SASP). Senescent cells are highly secretory and they execute a diverse set of functions mediated by SASP[17]. As senescent cells are viable and actively secreting cells, they have a profound influence on the surrounding cells. SASP affects senescent cells and their microenvironments in both autocrine and paracrine fashions. SASP factors are highly dynamic in expression and the composition changes over the period of senescence. This change in the composition of SASP in senescent cells may determine whether the effect is beneficial or harmful in the respective organs[17]. The mechanisms of the initiation and regulation of SASP and its influence in inflammation and disease processes have not been fully elucidated yet.
SIGNALING PATHWAYS IN SENESCENCE
The reactive oxygen species (ROS) is considered as one of the main activators of senescence cells[18,19]. The imbalanced ROS may cause oxidative stress and DNA damage, leading to the activation of DNA damage response in the affected cells.
Signaling pathways that control cell cycle arrest
The p53 and Rb signaling pathways may become activated in response to extrinsic and intrinsic stressors, including DNA damage. p53 and Rb can affect transcriptionally both upstream regulators and downstream effectors in the senescence pathway[20]. The inactivation of either p53 or Rb prevents or significantly delays senescence[21](Figure 1). ATM and ATR, as well as their downstream targets, Chk1 and Chk2, become activated during DNA damage, which in turn activate p53.Activated p53 is considered as one of the main players in converting normal cells to senescent cells. The phosphorylated form of p53 is the essential contributor of replicative senescence in human fibroblast[22]. Acetylation of p53 at Lys161/Lys162 is also important for cell cycle arrest and senescence[23]. Once p53 is activated by posttranslational modification, it in turn activates its transcriptional target, p21Cip1(CDKN1A). p21Cip1is the inhibitor of Cyclin E/CDK2 complex and it promotes cell cycle arrest at G1/S phase of cell cycle (Figure 1). Of note, p21Cip1can be activated by both p53-dependent and p53-independent mechanisms[24].
Retinoblastoma protein (Rb) is a tumor suppressor and a well-established cell cycle regulator. Rb is expressed in all tissues and controls cell cycle progression through interactions with the E2F family of transcription factors[20,25]. Other Rb family members, namely, p107 and p130, also play active and important role in driving senescence independently of Rb[26]. p21Cip1has been found to be both a positive and negative regulator of Rb by regulating either Rb phosphorylation or degradation,respectively[27]. p16INK4A(CDKN2A) inactivates Cyclin-dependent kinases, which phosphorylate Rb, and Rb phosphorylation status in turn has an impact on the expression of p16INK4A. In normal cells, Cyclin D and Cyclin E bind to CDK2 and CDK4/6, respectively, since p21Cip1and p16INK4Alevel remain at the basal level.Hyperphosphorylation of Rb permits the E2F factor to participate in the production of replication proteins and hence cell cycle progress. But a higher level of either p16INK4Aand/or p21Cip1can keep Rb in the hypophosphorylated state so that E2F remains bound with it. Therefore, cell cycle is arrested, which ultimately lead to cellular senescence (Figure 1).
Another cell cycle inhibitor, CDKN3 is a phosphatase and dephosphorylates Rb thus ensuring that cell cycle progression is blocked. Thus, CDKN3 is considered as an important contributor to cellular senescence[28]. CDKN3 can also interact with Mdm2 and form a complex with p53 and Mdm2. Once the complex is formed, p53 losses its ability to induce p21Cip1[29,30].
G1-phase cell cycle arrest can also be induced by transforming growth factor-beta(TGFβ)[31]. TGFβ can keep Rb in hypophosphorylated state, suggesting that it may suppress Rb phosphorylation and thereby interfering with cell cycle progression[32].p15INK4B(CDKN2B), a homologue of p16INK4A, is activated by TGFβ, acts on CyclinDCDK4/CDK6 complex and contributes to cell cycle arrest, and hence to senescence[33](Figure 1). Since p15INK4Band p16INK4Ahave a common mode of action, in a tissue where either one does not express, the other can possibly compensate the functionality.
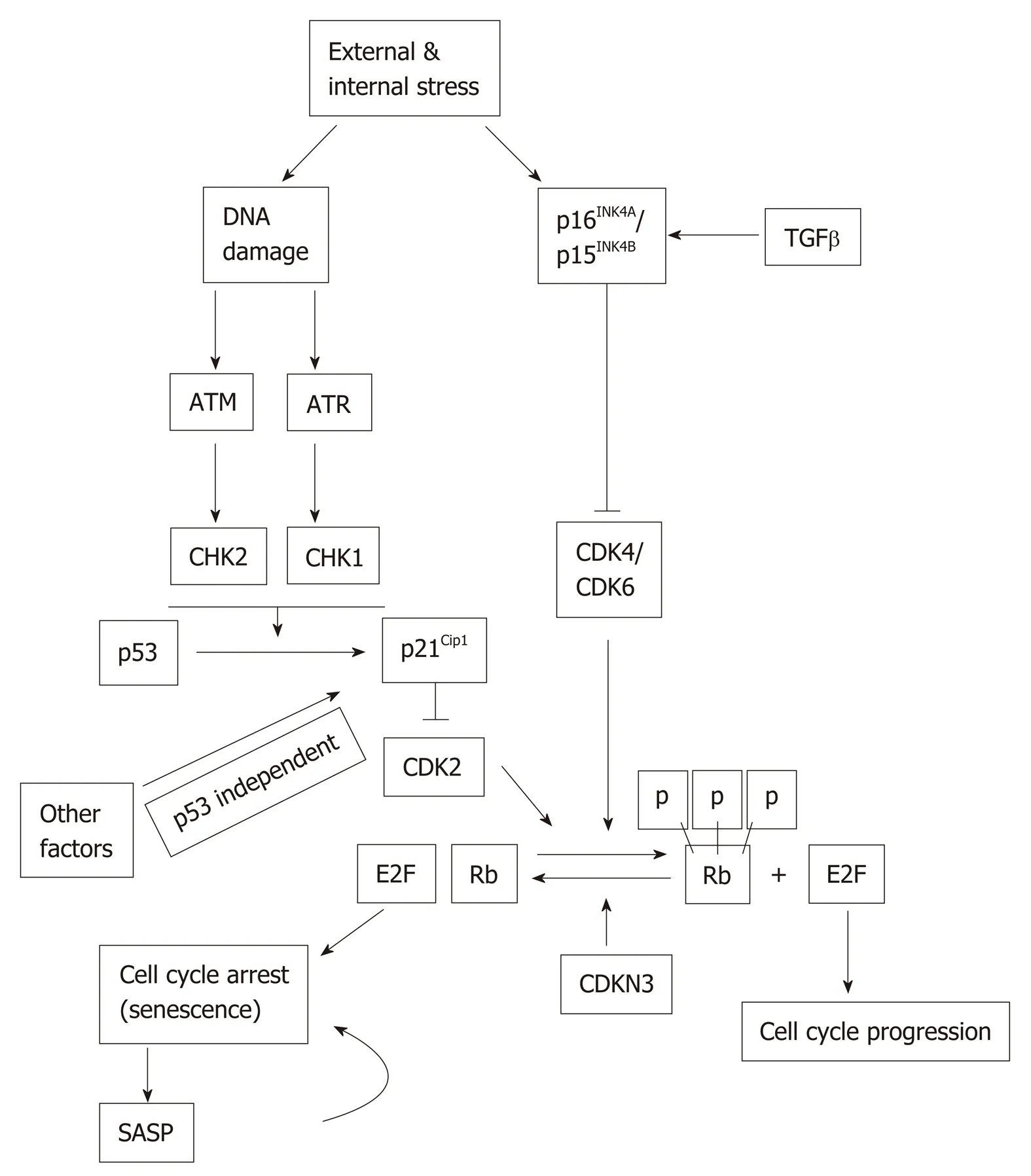
Figure 1 Stress-induced senescence. Both external and internal stresses can induce DNA damage and the activation of p16INK4A and/or p15INK4B. DNA damage can activate p53 ataxia telangiectasia mutated (ATM) and ATM and RAD3-related (ATR) pathway. Activated p53 induces p21 Cip1 expression. Expression of p21Cip1 can also be regulated by p53-independent mechanisms. The Cyclin dependent kinases activate Rb but are inhibited by p15INK4B/p16INK4A and p21Cip1 which leads to cell cycle arrest and senescence. The senescent cells express senescenceassociated secretory phenotype (SASP). The SASP factors may induce senescence in neighboring cells in a paracrine fashion. ATM: Ataxia telangiectasia mutated; SASP: Senescence-associated secretory phenotype.
Signaling pathways that regulate SASP
The connection between senescent cells and the immune system is mediated by SASP.The inflammation signature of SASP include chemokines, cytokines and other immune modulators. SASP is regulated at multiple levels’ such as, chromatin modification, transcription, translation, mRNA stability and secretion[34]. The autocrine and paracrine positive feedback loops also regulate the signaling of SASP.Most senescent cells express multiple cytokines such as IL-8, CCL2 (MCP-1), CCL7(MCP-3), CCL8 (MCP-2), CCL13 (MCP-4), CCL3, CCL16, CCL20, CXCL1, CXCL2,CXCL3ect[35,36]. The most characterized SASP components include multiple proinflammatory cytokines, such as interleukin-1 (IL-1), and interleukin-6 (IL-6). IL-1 and IL-6 produced in senescent epithelial and fibroblast cells are capable of inducing cellular senescence in adjacent cells. SASP helps spreading senescence phonotype among nonsenescent cells by creating an inflammatory environment in tissues. The knock-down of IL-6 and IL-8 receptors IL-6R and CXCR2, respectively, prevents senescence[34].
SASP is induced and regulated by several signaling pathways leading to the activation of the nuclear factor-κB (NF-κB) and/or CCAAT/enhancer-binding protein-β (C/EBPβ). In senescent cells, activated NF-κB and C/EBPβ regulate the expression of SASP factors by controlling, mainly at the transcription level, the inflammatory SASP molecules, IL-1α, IL-6 and IL-8. These inflammatory SASP factors can enhance SASP signaling through activating NF-κB , and C/EBPβ in an autocrine feed-forward fashion[37,38](Figure 2).
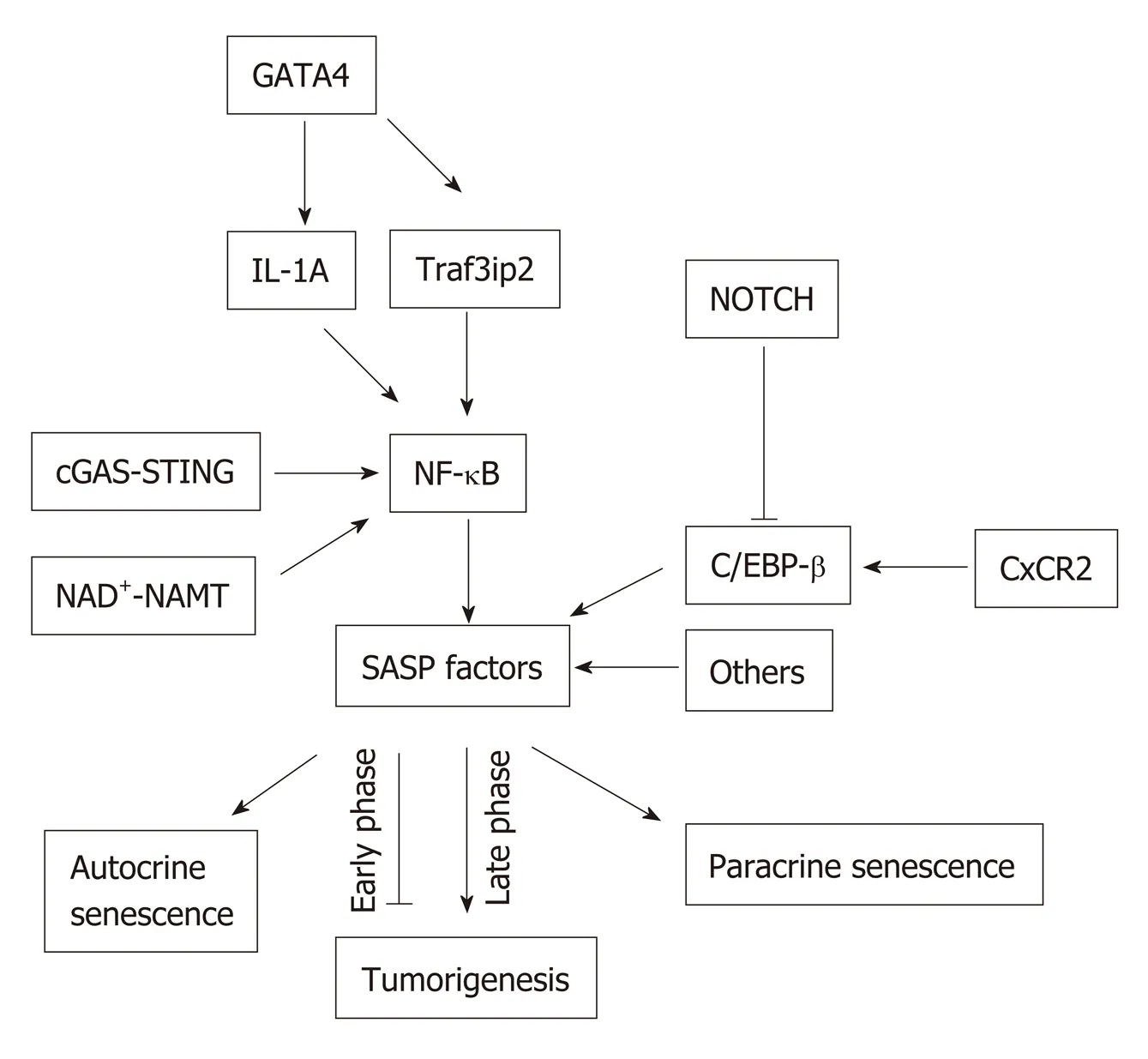
Figure 2 Senescence-associated secretory phenotype signaling pathways. Nuclear factor kappa light chain enhancer of activated B cells can be activated via multiple signaling pathways such as the GATA binding protein 4,cyclic GMP-AMP synthase-stimulator of interferon genes, and nicotinamide adenine dinucleotide -nicotinamide phosphoribosyltransferase NAD+-NAMT pathways, which lead to the expression of senescence-associated secretory phenotype (SASP) proteins. SASP can be positively regulated through C-X-C motif chemokine receptor 2, or negatively by NOTCH via CCAAT-enhancer-binding proteins. SASP can induce senescence in both autocrine and paracrine manners. SASP can be anti-tumorigenic in the early phase of senescence, but can be pro-tumorigenic in the late phase of senescence. NF-κB: Nuclear factor kappa light chain enhancer of activated B cells; GATA4: GATA binding protein 4; cGAS-STING: Cyclic GMP-AMP synthase-stimulator of interferon genes; SASP: Senescenceassociated secretory phenotype; CXCR2: C-X-C motif chemokine receptor 2; C/EBP: CCAAT-enhancer-binding protein.
NOTCH signaling pathway:This pathway has been implicated as an important regulator of SASP. Works from Hoareet al[39]suggested a global upregulation of NOTCH1 accompanied by dynamic alterations of its downstream activity in senescence. They also proposed NOTCH1 as a master regulator of SASP compositionviaa temporal and functional switch between two different secretomes, TGFβ and pro-inflammatory cytokines through down-regulation of C/EBPβ.
The NOTCH1 was shown to be upregulated in cells undergoing oncogene-induced senescence (OIS), which caused upregulation of TGFβ in the first phase of senescence.The inhibition of NOTCH signaling substantially reduced TGFβ induction in OIS cells. The inhibition of TGFβ in NOTCH1-driven senescent cells prevented the upregulation of the TGFβ-targeted cell cycle inhibitor, p15INK4B. In addition, Hoareet al[39]found that NOTCH1 negatively regulated C/EBPβ but had no effect on NF-κB pathway in the later phase of senescence. The overexpression of activated Notch1 receptors (N1ICD - NOTCH1 intracellular domain) inhibited the ability of C/EBPβ to induce IL-1, IL-6 and IL-8[39]. Moreover, Hoareet al[39]found that inhibiting Notch signaling accelerated clearance of senescent cells in the liver. Therefore, NOTCH signaling pathway may be associated with a complicated mechanism of SASP regulation. But the key questions that remain to be addressed are: (1) Which factor(s)involved in the switching from NOTCH1-TGFβ to NOTCH1-C/EBPβ; (2) What the cross-talk is between NOTCH1-mediated C/EBPβ and NF-kB regulation in senescent cells.
cGAS-STING pathway:cGAS (cGMP-AMP synthase) is a cytosolic DNA sensor and it is activated in response to cytosolic DNA. This activated cGAS binds and activates Stimulator of Interferon Gene (STING). During cellular senescence, the organization of chromatin changes and undergoes degeneration. The integrity of the nuclear envelope is disrupted due to the loss of the nuclear lamina protein Lamin B1.Eventually, small chromatin fragments migrate from nucleus to cytoplasm to become cytoplasmic chromatin fragments (CCF) in senescent cells[40]. cGAS can be activated by any double-stranded DNA irrespective of the sequence[41]. Therefore, CCF from the senescent cells can activate cGAS-STING pathway and upregulate SASP expression via inducing type-1 interferons (IFN-1). The cGAS-STING signaling pathway drives the production of inflammatory SASP cytokines through type I interferonviaIRF3 and proinflammatory responsesviaNF-κB[40,42], thereby facilitating senescence. Glucket al[43]demonstrated that cGAS deficiency in the liver abrogated the induction of senescence in hepatocytes, which suggests the importance of cGAS in the process of senescencein vivo.
Ceccoet al.[44]showed that in oncogene-induced senescence and stress-induced premature senescence, activation of retrotransposable element, L1, and IFN-1 occur in the late phase. They found three factors that caused L1 activation, RB1, FOXA1 and TREX1. RB1 expression was declined in senescent cells but was enhanced in proliferating cells. Upregulation of FOXA1 and downregulation of RB1 and TREX1 were found to be associated with L1 activation and stabilization. The activation of L1 ensured a strong activation of INF-1 response in senescent cells. In addition, when the cGAS-STING pathway was disrupted, the INF-1 response was inhibited and SASP response such as the induction of CCL2, IL-6, and MMP3 were downregulated in the late phase of senescent cells.
NAD+-NAMT pathway:NAD+and the rate-limiting enzyme of its synthesis,nicotinamide phosphoribosyltransferase (NAMPT), have a critical role in ageing and cancer[45-47]. Recently, Nacarelliet al[47]demonstrated a mechanism of SASP regulation that involves these factors and HMGA proteins HMGA1 and HMGA2[47]. HMGA proteins are known to regulate the chromatin structure and to promote senescence[48].Now, these proteins are found to regulateNAMPTgene expression by binding at the enhancer element during oncogene-induced senescence of fibroblasts[47]. NAMPT upregulates the expression of proinflammatory SASP fators such as IL-1β, IL-6 and IL-8, which are dependent on the enzymatic activity of NAMPT. This proinflammatory activity are regulated by increased NAD+/NADH ratio. Nacarelliet al[47]have also demonstrated that increased NAD+/NADH ratio causes suppression of AMPK, which interferes with p53-mediated inhibition of p38 MAPK. p38 MAPK controls the transcriptional activity of NF-κB by regulating acetylation of p65. The NF-κB mediated proinflammatory activity is therefore enhanced.
In addition to HMGA proteins, another high mobility group protein, HMGB1, has been implicated in the senescence and SASP. Davaloset al[49]reported the existence of a relationship between HMGB1 and senescence. They found that altered expression of HMGB1 induced senescence in a p53-dependent manner in fibroblasts. Exogenous HMGB1 treatment activated NF-κB activity and IL-6 secretion whereas depletion of HMGB1 attenuated the senescence phenotype. The disruption of normal HMGB1 levels can induce a p53-dependent cell cycle arrest. Interestingly, secreted HMGB1 is essential for optimal secretion of IL-6 and MMP-3, both are important SASP components.
Other pathways:The other known regulators of SASP are bromodomain containg protein 4 (BRD4), lysine methyltransferase MLL1 and G9A[50]. The recruitment of BRD4 to senescence-activated enhancers located adjacent to SASP genes is probably required to induce SASP[51]. But the upstream signaling that regulate these transcriptional activators are not completely known. SASP helps spreading senescence phonotype among nonsenescent cells by creating an inflammatory environment in tissues.
For SASP regulation, mTOR plays an important role as well. The 4E-BP1 is phosphorylated by mTOR. It is a repressor of translation of mRNA. In order to regulate SASP, 4E-BP1 represses IL-1α and MAP kinase-activated protein kinase 2[52].The degradation of mRNA of proinflammatory SASP factors is associated with a RNA binding protein, zinc finger protein 36L1 (ZFP36L1); and MAP kinase-activated protein kinase 2 inhibits ZFP36L1[34]. By regulating the stability of SASP mRNA,mTOR regulates SASP in senescent cells.
Autophagy and senescence
Autophagy is a highly regulated cellular program responsible for recycling intracellular proteins and damaged/nonfunctional organelles. Autophagy was found to be activated in senescence in human fibroblasts due to the negative feedback of mTOR pathway[53]. A number of genes (e.g., Ulk3 and LC3) associated with autophagy are upregulated in senescent cells and inhibition of autophagy essential genes like Atg5 or Atg7 caused interference of senescence phenotype including SASP[53].Reciprocally, Kanget al[54]showed that knockdown of autophagy essential genes such as Atg7, Atg12 or lysosomal associated membrane protein 2 (LAMP2) caused senescence in human fibroblasts. This senescence pathway was found to be ROS and p53-dependent. In contrast, Garcia-Pratet al[55]demonstrated that muscle stem cells maintain a reversible quiescence state and do not enter to senescence when the autophagy mechanism is active. This group showed that the muscle stem cells underwent senescence if autophagy was defective and restoration of autophagy prevented senescence in satellite cells. Therefore, autophagy may both induce and prevent senescence, which may be dependent on cell type or the type of experimental model. However, it is still an open question as to whether autophagy is necessary for senescence to occur or it inhibits the senescence process.
Kanget al[56]reported that a transcription factor, GATA4 played an important role in the cellular senescence mechanism. This group suggests that GATA4 is a senescence and SASP regulator. In normal cells, GATA4 binds to an autophagy adaptor, p62, whichis eliminated by the selective autophagy process. This autophagy process is suppressed when senescence is induced. DNA damage activates ATM and ATR kinases, which inhibit p62-mediated GATA4 degradation. The stabilized GATA4 then activates NF-κB inducers TRAF3IP2 and IL-1A. NF-κB, one of the master regulators of senescence, initiates and maintains SASP, and facilitates senescence. The GATA4 activation is found to be independent of p53 and p16INK4Abut is dependent on ATM and ATR kinases. This finding suggests that GATA4 may represent a separate branch of DNA damage repair response that induces SASP and hence favors senescence. Kanget al[56]has suggested a clarification on the argument whether autophagy induces or inhibits senescence. Selective autophagy prevents cells from undergoing senescence by limiting GATA4 level when senescence-inducing stimuli,such as irradiation are given but later general autophagy lead to cellular senescence[57].
At least, one more member of the GATA family, GATA6, has been implicated in the senescence mechanism. Perlmanet al[58]reported that GATA6 overexpression could upregulate the expression of p21Cip1so that the proliferation of vascular smooth muscle cells and fibroblasts were inhibited. Zhanget al[59]suggested the existence of a direct relationship between autophagy and GATA6. Autophagy decreased accumulation of GATA6 and p62 was found to be a negative regulator of GATA6 accumulation. Zhanget al[59]also determined the relationship between an antimalarial drug, dihydroartemisinin (DHA) and GATA6, and their contribution to the hepatic senescence. DHA treatment enhanced the expression of p53, p21Cip1and p16INK4Aand induced senescence in hepatic stellate cells (HSCs) in rat livers. GATA6 accumulation further accelerated the expression of p53 and p16INK4Ain DHA-treated livers and promoted senescence. The knock-down of GATA6 by siRNA significantly reduced DHA-induced upregulation of p53 and p16INK4A, and thereby the senescence.
SENESCENCE IN THE LIVER
The detailed mechanism and biological function of cellular senescence in liver diseases have not been fully elucidated. But meaningful progresses have been made in this field. Senescence can adversely affect liver functions, cellular viability and tissue regeneration under pathological conditions[60]. Senescence in hepatocytes is well documented in different liver diseases such as cirrhosis[61,62]. Senescence in cholangiocytes[60,63,64]is also known to occur in such biliary diseases as primary biliary cholangitis (PBC) and primary sclerosing cholangitis (PSC). Senescence in HSCs has been reported in fibrosis[65].
Mouse models to study liver cell senescence
There are various mouse models developed to study senescence-associated liver diseases. Demariaet al[66]developed a mouse model named p16INK4A-3MRto visualize and to eliminate senescent cellsin vivo. The promoter of p16INK4Awas used in this model to express a fusion protein, composed of luciferase, red fluorescence protein(RFP) and herpes simplex virus 1 thymidine kinase. This viral thymidine kinase converts ganciclovir (GCV) into an apoptosis inducer. GCV treatment induces p53 accumulation and surges cell surface expression of CD95 and tumor necrosis factor receptor. Utilizing the p16INK4A-3MRmice and imaging, researchers can investigate a vigorous, but transient, upsurge in senescent cellsin vivo.
AhCre+Mdm2fl/flmice[9]: Murine Double Minute 2 (MDM2) interferes the transcriptional activity of p53 and guides degradation of p53 through ubiquitination.The disruption of the p53-MDM2 interaction by knocking out MDM2 genetically leads to p53-mediated cell-cycle arrest through inducing the activation of cell cycle inhibitor, p21Cip1[67]. Deletion of Mdm2 causes upregulation of p53 and thus p21Cip1.The AhCre system allows expression of Cre recombinase in hepatocytes in response to β-naphthoflavin, which in turn deletes the floxed Mdm2. This AhCre+Mdm2fl/flmouse model induces p21Cip1-dependent hepatocellular senescence conditionally[9]. The rescue of growth arrest may be achieved by crossing the AhCre+Mdm2fl/flmouse strain to p21Cip1-deficient mouse strain.
K19-Mdm2f/ftdTomLSL[60]: In this mouse model, Mdm2 can be conditionally deleted in cholangiocytes as the Cre recombinase expression is under the control of Krt19 promoter. This model exhibits features of biliary diseases upon induction of senescence in cholangiocytes. The Cre activity in senescent cholangiocytes can be monitored following the expression of tdTomato[60].
Mdr2(Abcb4)(-/-) mouse model is useful for the study of chronic biliary liver disease. Themdr2gene encodes a membrane protein that transports phosphatidylcholine.mdr2-deficient mice are unable to secrete phosphatidylcholine into the bile by hepatocytes which leads to cholestatic liver disease. Researchers have used this model to study senescence in cholangiocytes and fibrosis in the liver[68-70].
Senescence-accelerated mouse prone8 (SAMP8) is widely studied for aging mechanism, but is also useful for the study of liver diseases because of its phenotype of liver dysfunction[71]. The SAMP8 mouse strain exhibits an oxidative stress-induced age-related phenotype and severe mitochondrial dysfunction related liver pathology[72].
Senescence in hepatocytes
The function and the regenerative capacity of the liver deteriorate with age[73]. The accumulation of reactive oxygen species (ROS) during aging could lead to a significant level of oxidative stress. A number of studies have suggested a causative connection between cellular injury and oxidative stress in aged individuals[74,75]. Intanoet al[76]reported that DNA damage could be caused by ROS and the repair response was slowed down in aged mouse hepatocytes. ROS and DNA damage trigger cells to overexpress cell cycle inhibitors and halt proliferation of the damaged cells by inducing senescence. The liver can repair and regenerate normally if the damage is mild. But when the degree of damage to the liver is severe, hepatocytes lose regenerative capacity, and undergo necrosis, apoptosis or senescence[11,77].
Hepatocellular senescence can cause remarkable changes in tissue homeostasis, and microenvironment through SASP, which may lead to fibrosis and hepatocellular carcinoma. In the early stage of liver diseases, hepatocellular senescence may serve as a protective role by blocking proliferation of affected or injured liver cells. Therefore,the risk for these affected cells to be transformed to cancer cells would be reduced. But at the later stage of the diseases, senescent cells secrete SASP factors with an abnormal level of proinflammatory cytokine/chemokines, such as CCL2, and TGFβ, which can promote the conversion of non-senescent cells to senescent cells in a paracrine manner. Rudolph and DePinho have hypothesized that chronic liver injury causes progressive and repetitive liver damage and regeneration, which leads to accumulation of shorter telomeres in hepatocytes. This in turn may lead to hepatocellular senescence and ultimately cirrhosis[78]. In addition, Wiemannet al[62]showed that telomere shortening and hepatocellular senescence were positively correlated with progression of fibrosis in human cirrhosis samples, which denoted hepatic senescence as a general marker of cirrhosis.
The accumulation of fat in hepatocytes can promote telomere shortening and DNA damage, which may be mediated by oxidative stress. The critically shorter telomere and damaged DNA may induce senescence in hepatocytes[11,14]. Aravinthanet al[14]demonstrated that hepatocytes in non-alcohol-related fatty liver disease (NAFLD)could enter the senescent state after they overexpressed p21Cip1, and presented a larger nuclear area and γ-H2AX positive foci. The presence of hepatocyte senescence positively correlated not only with the fibrotic stage of the liver but also with the diabetic state associated with NAFLD.
Any repetitive wave of insult that can cause damage to hepatocytes such as alcohol intake, hepatitis viral infection, immune disorder or autophagy deficiency, may compromise liver regeneration and promote senescence in hepatocytes and other types of cells[64]. The normal liver contains a basal level of senescent hepatocytes (3%-7%), but in chronic hepatitis and cirrhosis the liver may exhibit 50%-100% of hepatocytes in senescence[61,62]. The generation of fibrotic scars in cirrhosis is thought to be the consequence of hepatocyte senescence, which may be used as a marker for cirrhosis[62].
Teohet al[79]reported the induction of p53, p21Cip1and p16INK4Ain senescent hepatocytes during chronic liver diseases but the detailed mechanism was not studied. Recently Birdet al[9]published a seminal article showing that acute liver injury could also induce hepatocyte senescence. Using a hepatocyte specific senescence induction model (AhCre-Mdm2fl/fl) they found that hepatocytes undergoing cell-autonomous senescence can transfer senescence properties to normal hepatocytesviaa SASP factor, CCL2, which stimulates the production of TGFβ from hepatic macrophages.
The TGFβ pathway was found to be activated in senescent hepatocytes in human fulminant liver failure. The targets of TGFβ pathway, SMAD7 and pSMAD2/3, were upregulated in hepatocytes near the necrotic area during acute liver injury. The interference of monocyte recruitment with an anti-CCL2 antibody in this model led to reduction of senescence spreading from senescent hepatocytes to normal hepatocytes,and enhanced hepatocyte proliferation. This finding suggests that TGFβ and CCL2 can work together in a non-cell-autonomous senescence pathway. The inhibition of TGFβ-TGFβ-R1 interaction by specific inhibitors or the depletion of macrophages by Liposomal Clodronate reduced senescence progress and enhanced liver regeneration[9]. This study thus suggests that the senescence process in acute liver injury may be therapeutically manageable.
Senescent hepatocytes can recruit and activate immune cells using their SASP property. The activated immune cells can remove senescent hepatocytes, which is termed as ‘‘senescence surveillance’’, hence preventing malignant transformation[80,81].Hepatic senescence harboring additional mutations like, p53 mutation, can cause aggressive hepatocellular carcinoma (HCC)[82]. Restoration of p53 in liver tumors can activate immune cell-mediated clearance of senescent tumor cells[83,84]. Kanget al[80]found that CD4+T cells clear senescent hepatocytes with the help of activated monocytes/ macrophages, which also suggested to the importance of senescence surveillance as an antitumor barrier for the HCC.
Senescence in cholangiocytes
The existence of senescent cholangiocytes and their potential deleterious effects in biliary diseases (PBC and PSC) have been described[85,86]. To have a better understanding of the detailed mechanism of cellular senescence in cholangiopathies,anin vivomodel (K19-Cre-Mdm2f/f)[60]was recently created to study the mechanism of cholangiocyte mediated biliary senescence in the liver by conditionally activating senescence in cholangiocytes. The cell-autonomous senescence, where the induction of p21Cip1and p16INK4Aare dependent on p53 induction in cholangiocytes, results in the conversion of normal hepatocytes of close proximity into non-cell-autonomous senescence, where the induction of p21Cip1and p16INK4Aare independent on p53[60]. As in the AhCre-Mdm2fl/flmodel this paracrine senescence was found to be TGFβdependent as the blocking of TGFβ signaling pathway partially interfered with the paracrine senescence effect. The involvement of other SASP factors in addition to TGFβ may need to be explored in additional details.
Both studies by Ferreira-Gonzalezet al[60]and Birdet al[9]have indicated the importance of TGFβ in the transmission of senescence properties from senescent cells to otherwise normal cells. TGFβ has been shown to be involved in the liver disease progression from injury to hepatocellular carcinoma[87]. Therefore, TGFβviasmall molecule inhibitors could be a valuable therapeutic target to treat cholangiopathies and hepatocellular injury and preventing senescence-associated tumorigenesis.
In cholestatic liver diseases and cholangiopathies, the bile acid (BA) secretion can be defective and bile acids may not reach to intestine, which may cause accumulation of bile acids in the liver, and cholestatic liver injury, which in turn can induce senescence in liver cells[88,89]. Whether a specific BA species can directly cause senescence is not known.In vitrotreatment of bile duct epithelial cells with deoxycholic acid (DCA) and lithocholic acid (LCA) could not induce SASP. BA may induce senescence in other cells, such as HSCs.
Senescence in HSC
HSCs are in quiescent state in normal healthy livers but become activated following liver injury. Activated HSC play an important role in liver fibrosis. There are three possible fates for the activated and fibrogenic HSC: (1) Apoptosis, to limit the HSC involvement in extracellular matrix (ECM) deposition; (2) Reversal from the activated status to quiescence; and (3) Senescence, to limit the degree of fibrosis in the liver.Multiple inducers of senescence in HSC have been reported, including IL-22, CCN1(CYR61), retinoic acid, and Substance P ( SP)[69,90].
Hepatocellular fibrosis often results from the excessive deposition of ECM by activated stellate cells. The general concept is that senescent hepatocytes are associated with fibrosis, impaired liver function and increased mortality. On the other hand, Krizhanovskyet al[65]reported that in response to liver injury, activated HSC initially proliferated and contributed to ECM deposition. However, senescent HSC could limit the extent of fibrosis. Moreover, they found that if HSCs were deficient in key senescent genes in the p53 or Rb pathways, these cells continued to proliferate and secreted excessive ECM. These finding suggest that senescent HSCs are probably beneficial for the injured liver but the senescent hepatocytes are not.
A secondary bile acid, DCA can induce SASP in HSC[91]. The enterohepatic circulation of DCA seemed important for the senescence of HSC. DCA can induce DNA damage by upregulating ROS, which may lead to senescence in HSC but the underlying mechanism is unclear. It is not known if the bile acids can induce senescence in other liver cells.
When HSC proliferate, they secrete molecules that form ECM such as collagens(e.g., Col1A) and matrix metalloproteins (MMPs). In acute liver injury, senescent HSC may have antifibrotic effects by secreting matrix metalloprotenases that digest MMPs and collagens. However, senescent cells of other origins (non-HSC) can express matrix metalloprotenase inhibitors, which would promote fibrosis by antagonizing the effect of senescent HSC. Therefore, detailed studies are required to understand the balance between fibrogenic and non-fibrogenic SASP in senescent HSC and in senescent non-HSC.
CONCLUSION
An increasing number of research on hepatocellular senescence have indicated the importance and significance of the senescence in the liver. The detailed mechanisms of hepatic senescence are yet to be fully determined. Exploring the regulatory mechanisms of SASP discussed above in the liver will provide an important insight to fight against hepatic inflammation, fibrosis or cancer in the liver tissues.Understanding the molecular mechanisms of cellular senescence in liver diseases will help to develop important tools for better diagnosis and treatment of the liver diseases.
杂志排行

World Journal of Gastroenterology的其它文章
- Diagnostic and prognostic potential of tissue and circulating long non-coding RNAs in colorectal tumors
- Autoantibodies: Potential clinical applications in early detection of esophageal squamous cell carcinoma and esophagogastric junction adenocarcinoma
- Role of endoscopic ultrasound in the screening and follow-up of high-risk individuals for familial pancreatic cancer
- Optimizing proton pump inhibitors in Helicobacter pylori treatment:Old and new tricks to improve effectiveness
- Regulatory effect of a Chinese herbal medicine formula on nonalcoholic fatty liver disease
- Allyl isothiocyanate ameliorates lipid accumulation and inflammation in nonalcoholic fatty liver disease via the Sirt1/AMPK and NF-κB signaling pathways