Diagnostic and prognostic potential of tissue and circulating long non-coding RNAs in colorectal tumors
2019-09-25OrsolyaGalambBarbaraBartAlexandraKalmZsfiaNagyKrisztinaSzigetiZsoltTulassayPeterIgazlaMoln
Orsolya Galamb, Barbara K Barták, Alexandra Kalmár, Zsófia B Nagy, Krisztina A Szigeti, Zsolt Tulassay,Peter Igaz, Béla Molnár
Abstract Long non-coding RNAs (lncRNAs) are members of the non-protein coding RNA family longer than 200 nucleotides. They participate in the regulation of gene and protein expression influencing apoptosis, cell proliferation and immune responses, thereby playing a critical role in the development and progression of various cancers, including colorectal cancer (CRC). As CRC is one of the most frequently diagnosed malignancies worldwide with high mortality, its screening and early detection are crucial, so the identification of disease-specific biomarkers is necessary. LncRNAs are promising candidates as they are involved in carcinogenesis, and certain lncRNAs (e.g., CCAT1, CRNDE, CRCAL1-4) show altered expression in adenomas, making them potential early diagnostic markers.In addition to being useful as tissue-specific markers, analysis of circulating lncRNAs (e.g., CCAT1, CCAT2, BLACAT1, CRNDE, NEAT1, UCA1) in peripheral blood offers the possibility to establish minimally invasive, liquid biopsy-based diagnostic tests. This review article aims to describe the origin,structure, and functions of lncRNAs and to discuss their contribution to CRC development. Moreover, our purpose is to summarise lncRNAs showing altered expression levels during tumor formation in both colon tissue and plasma/serum samples and to demonstrate their clinical implications as diagnostic or prognostic biomarkers for CRC.
Key words: Long non-coding RNA; Colorectal cancer; Colorectal adenoma; Circulating long non-coding RNAs; Exosome; Biomarker; Diagnostic marker; Prognostic marker
INTRODUCTION
Colorectal cancer (CRC) is one of the most frequent malignant diseases worldwide with a remarkably high mortality rate[1]. The number of CRC-related deaths can be reduced only by diagnosis at the earliest stage when the disease is more likely to be cured.
Long non-coding RNAs (lncRNAs), a novel family of non-protein coding RNAs(200 nt-10 kb) are of outstanding interest as their expression is often altered in various disease types including malignancies[2]. They are known to have a crucial role in the regulation of gene expression, alternative splicing mechanisms, protein localization and activity, formation of cellular substructures and protein complexes through their diverse interactions with DNA, RNA and proteins[3,4].
In cancers, lncRNAs are involved in every stage of carcinogenesis and tumor progression including tumor initiation, proliferation, apoptosis and migration of cancer cells, angiogenesis, tumor invasion and metastasis formation[5,6]. Their altered expression can influence several oncogenic signaling cascades including the WNT/βcatenin, PI3K/Akt, EGFR, NOTCH, mTOR and TP53 signaling pathways[4,7-20]. Besides local expression changes in cancerous tissue and tumor-related stroma, lncRNAs also remain stable in body fluids due to their resistance to RNases[2,21].
Several lncRNAs showing altered expression in colorectal tumors including precancerous adenomas have potential as early diagnostic markers[22-24]. In this review,we summarize the colorectal tumor-related tissue and circulating lncRNAs, altered lncRNA expression patterns, and technical aspects of their isolation and detection.Our aim is to show their potential as diagnostic and prognostic biomarkers based on recently published data.
HISTORY, CLASSIFICATION, FUNCTION, LIFETIME AND SUBCELLULAR LOCALIZATION OF LNCRNAS
Regulatory non-coding RNAs (ncRNAs) were first reported in eukaryotes in the 1980s, of which H19[25]and Xist[26]were the first members of the family[27]. When the Human Genome Project was completed, it became clear that only a minor part of our genome codes proteins and the rest was considered as “junk” DNA[28]. Since then, our knowledge about the non-coding genome was expanded, and the still unexplored regulatory role of the ncRNA world is the focus of several studies and holds a significant clinical potential[29]. Over the past decades, along with the development of explorative molecular biology methods, the importance and function of the complex eukaryotic transcriptome have been recognized, a large proportion of which comprises the actively transcribed lncRNAs[30]. After the discovery and the intensive analysis of the class of small ncRNAs called miRNAs since 1993[31], it became evident,that other ncRNAs also play fundamental role in gene expression regulation, and that their alterations can be responsible for the disrupted molecular pathways in multiple cancers[32].
The major class of ncRNAs are lncRNAs, which are derived from highly diverse genomic context and are classified on the basis of the genomic region of origin[28].According to the genomic database [Ensembl Release 96 (April 2019)], human lncRNAs are categorized into 3prime overlapping ncRNA, antisense, lincRNA (long interspersed ncRNA), retained intron, sense intronic, sense overlapping and macro lncRNAs. The lncRNAs that are not overlapping with protein-coding genes are called stand-alone lncRNAs including the large intergenic (or intervening) lncRNA(lincRNA) group[28,33](e.g., XIST, H19, MALAT1, and HOTAIR). Antisense lncRNAs are transcripts overlapping the genomic strand of a protein-coding locus in an antisense direction[34], while sense lncRNAs are overlapped with the sense strand of protein coding genes containing exons[35]. Antisense transcription is widespread in the mammalian genome[36]; the estimated ratio of the genes with antisense transcripts varies from less than 2 to more than 70% of the total genes[37]. XIST/TSIX is a wellknown example of the sense-antisense transcript pairs[38]. Pseudogenes are defined as nonfunctional sequences of genomic DNA originally derived from functional genes[39].Long intronic ncRNAs are transcribed from the intronic sequence of a coding gene.On the basis of their association with functional DNA elements, enhancer- and promoter-associated lncRNAs can be distinguished[40]. The lncRNAs localize in the cytoplasm, nucleus, nucleolus, and also in other subcellular compartments and vesicles (such as nuclear bodies, exosomes) and the localization is related to their molecular functions[41]. Certain sequence motifs in their primary sequence are associated with the subcellular localization[42].
As the largest class of non-coding transcripts, lncRNAs have a wide variety of functions. They can act as RNAs (e.g., ribozymes, riboswitches)[43]and widely as ribonucleoprotein particles (RNP)[44]. They can exert their positive or negative regulating functions eitherin cisorin trans[45]. One of their functions is the regulation of nuclear organization; lncRNAs can modulate the chromatin architecture (e.g., Xist)and they can also regulate inter- and intrachromosomal interactions (e.g., colorectal cancer associated transcript 1. long isoform (CCAT1-L) modulating interchromatin loops between enhancers and promoters[46]). LncRNAs can regulate other non-coding RNAs (e.g., as miRNA sponges leading to reduced miRNA inhibitory effect on target molecules[47]), and also can be processed into single- or double-stranded siRNAs[3].Several gene transcription processes can be activated or blocked by lncRNAs by recruiting or inhibiting transcription factors of the target gene promoters[3,44]. Certain lncRNAs are linked to the process of alternative splicing (e.g., LINC001133)[48].Furthermore, protein activity is regulated by lncRNAs and trafficking between the subcellular compartments can also be influenced by lncRNAs[49].
Nuclear lncRNAs also contribute to chromatin remodeling as they can promote or prevent the recruitment of chromatin modifiers[46]. They are also part of nuclear bodies[50]with scaffold function, so-called architectural lncRNAs[51][such as nuclear enriched abundant transcript 1 (NEAT1), as a well-characterized lncRNA as a crucial component of paraspeckles[52]] and also as non-architectural lncRNAs [e.g., metastasis associated lung carcinoma transcript 1 (MALAT1) as one of the most abundant lncRNA in nuclear speckles[46]].
Epigenetic mechanisms, such as histone modifications are also influenced by lncRNAs. For instance, lncRNA HOTAIR (homeobox transcript antisense intergenic RNA) interacts with both LSD1/CoREST/REST complex and PRC2 as a modular scaffold that leads to coupled histone H3 lysine 27 methylation and lysine 4 demethylation[53].
By the modulation of all three major mammalian DNA methyltransferases(DNMT1, DNMT3a, DNMT3b), lncRNAs influence DNA methylation levels resulting in altered expression of the target genes[44]. DNMT1-associated Colon Cancer Repressed lncRNA 1 (DACOR1) interacts with both chromatin and DNMT1 and targets DNMT1 protein complex to certain genomic loci, also affecting cellular SAM levels[54,55]. Altogether, the expression alterations of lncRNAs influence many biological functions that contribute to the disturbance of the complex fine-tuning machinery of non-coding RNA regulatory network during cancer formation.
Our knowledge about the posttranscriptional regulation of lncRNAs is limited,however, the stability of transcripts can be an important aspect in gene expression regulation[56,57]as the half-life of ncRNAs correlates with their functional characteristics[58]. Each lncRNA has a unique structure, and these transcripts are characterized by complex secondary and tertiary structures which is crucial to exert their functions[59]. Although the stability of these non-coding transcripts was generally considered to be lower compared to mRNAs[60]on the basis of a genome-wide lncRNA analysis by Clarket al[56], a wide variety in their stability can be observed which is consistent with their functional diversity. LncRNA stability is correlated with genomic location, subcellular localization, splicing, and GC percentage, while in contrast,expression levels are not correlated with stability[56]. The half-life of lncRNAs ranges from < 30 min to > 48 h with median value at 3.5 h, and they can be classified as unstable and to highly stable lncRNAs - the latter represented at a lower percentage[56]. According to Clarket al., nuclear-enriched lncRNAs displayed significantly lower stability compared to those detected both in nucleus and cytoplasm[56]. It is important to note that lncRNAs with even lower stability have been shown to have fundamental role (e.g., NEAT1 as scaffold lncRNA of paraspeckles, as dynamic nuclear subdomains[61]), furthermore, the existence of highly stable lncRNAs illustrate the biomarker potential of this subclass of non-coding transcripts.
LNCRNA EXPRESSION ANALYSIS METHODS
Analysis of lncRNAs is technically challenging due to their relatively low expression level and their tissue-specific expression[62], therefore, the following methods are optimized for studying lncRNAs with high sensitivity and resolution.
High-throughput sequencing serial analysis of gene expression (SAGE) is based on short cDNA sequences containing recognition sites for restriction enzymes at the transcripts’ 3’ end, and it was one of the first transcriptome analysis methods to study lncRNA expression[63,64].
Among whole genome analyses, microarrays are widely used to analyse the RNA expression in a high-throughput manner from the 2000s, however, these systems are limited to studying the known RNAs. Furthermore, cross-hybridization and limited detection range due to background and saturation signals make these analyses more challenging[65]. In parallel, the rapid development of next generation sequencing(NGS) systems revolutionized the experimental field, as RNA-Seq provides a costeffective and rapid solution for whole transcriptome profiling with the potential to discover novel transcripts[65]. The higher resolution and reproducibility of RNA-Seq compared to microarrays[65]resulted in broad use of this approach. RNA-Seq supports the annotation of novel lncRNAs, RNA editing sites, and alternative splicing sites, as well[62]. Cap analysis of gene expression (CAGE) is an NGS-based approach to map and quantify the expression of 5’ capped RNAs[66]and also to identify transcriptionally active promoter regions and Pol II-driven TSSs[64].
The lncRNAs regulate and mediate interactions on different molecular levels and complex networks of these non-coding RNAs remain to be explored. RNA-binding protein immunoprecipitation (RIP) is used to study RNA-protein interactions, where the RNA of interest can be complexed with its interacting proteins, and this fraction can be selectively pulled down[67]. The downstream analysis can be performed by combining with the previously discussed methods, including RIP-Chip and RIPSeq[68]. Native RIP is suitable for the exploration of strong and direct RNA-protein interactions, whereas the crosslinked immunoprecipitation method (CLIP) is used to study weak or indirect binding[62]. Crosslinking is achieved by ultraviolet light (UV)followed by RNase treatment and stringent washes which increases the specificity of the interaction detection[69]. In order to minimize the disadvantages of CLIP, modified methods, such as individual nucleotide resolution CLIP (iCLIP)[70], and photoactivable ribonucleoside-enhanced CLIP (PAR-CLIP) are also available for the identification of the exact crosslinking sites with single nucleotide resolution[62,69].
Other RNA pull-down methods, such as chromatin isolation by RNA purification(ChIRP)[71], capture hybridization analysis of RNA targets (CHART)[72]and RNA antisense purification (RAP)[73]can be applied to study RNA-DNA interactions to shed light on lncRNAs’ functions and identify trans-genomic interacting sites[62]. During ChIRP experiments, a biotin-labeled antisense probe designed to the selected lncRNA is employed to explore its interacting chromosomal fragments[71]. Different probe design criteria are applied in the case of CHART, as in contrast with ChIRP probes spanning the whole interesting lncRNA, the CHART method uses capture oligos specific for the accessible regions of the lncRNA candidate[72]. The co-purified RNA,DNA or proteins potentially interacting with the selected lncRNA can be analysed with NGS, PCR or Western blotting[62]. RAP can be performed with different crosslinking methods (e.g., psoralens) along with the longer biotinylated probes (> 60 bp) to enhance the RNA-DNA hybrid stability[73]and to reduce the signal-to-noise ratio[64].
LncRNAs are known to exert their function also by binding directly or indirectly to other RNAs[64]. These interactions can be studied by RAP-RNA (applying different chemical cross-linking), as 4’aminomethyltrioxalen: RAP-RNA[AMT], formaldehyde:RAP-RNA[FA], FA and disuccinimidyl glutarate: RAP-RNA[FA-DSG][74]or UV-crosslinked CLASH (cross-linking, ligation and sequencing of hybrids)[75]methods.
It is known that lncRNAs fold into secondary and tertiary structures that are crucial to exert their regulatory effects[59], but the structural domains of the RNA interactome still need to be explored. Structural relationships can be studied by dimethyl sulfate sequencing (DMS-Seq), selective 2’-hydroxyl acylation analysed by primer extension sequencing (SHAPE-Seq), genome-wide fragmentation sequencing (FRAG-Seq), and parallel analysis of RNA structure (PARS) techniques[76]. By the intensive development of subcellular visualization approaches, lncRNAs can be localized within the cell with high sensitivity using special fluorescentin situhybridization (FISH) applications(single molecule FISH - smFISH, sequential FISH - seqFISH, and multiplexed errorrobust FISH - MerFISH)[77-79]. High resolution microscopes, as structured illumination microscopy (SIM[80]) or stochastic optical reconstruction microscopy (STORM[81])enable the precise detection of certain lncRNAs and investigation of their colocalization partners[46]. The functional investigations of lncRNAs can be performed with antisense oligonucleotides (ASO) and also by siRNAs and shRNAsviabinding and affecting the target lncRNA’s functionality[82]. The CRISPR-Cas9 genome editing technique[83]has revolutionized functional studies in the lncRNA world, which can be employed to silence (CRISPRi[84]) and also to overexpress (CRISPRa[85]) the lncRNA of interest[86].
LNCRNA EXPRESSION ALTERATIONS IN COLORECTAL ADENOMA AND CANCER TISSUE
Increasing evidence suggest that lncRNAs are involved in the whole process of CRC development, progression and metastasis formation - similarly to their diverse regulatory role in other types of malignancies - affecting the essential signaling pathways including WNT, TP53, PI3K/Akt, mTOR, EGFR and NOTCH1 in CRC[4-20].Abnormal expression of numerous lncRNAs including the well-known HOTAIR[87-90],MALAT1[91-93]and H19[94,95]has been described in CRC compared to normal colonic tissue samples (Table 1). From a clinical point of view, lncRNAs - with altered expression in different stages of colorectal carcinogenesis, and disease progression -have a particularly great potential to become early diagnostic and/or prognostic biomarkers.
Several studies reported the altered expression of certain lncRNAs including colon cancer associated transcript-1 (CCAT1), colorectal neoplasia differentially expressed(CRNDE-L), colorectal cancer associated lncRNA (CRCAL) 1, -2, -3 and -4 and urothelial carcinoma-associated 1 (UCA1) already in precancerous adenomas[23,24,37,96-102].
Nissanet al[96]in their comprehensive RT-qPCR study were the first to demonstrate the massive (often more than 100-fold) upregulation of CCAT1 in CRC and premalignant adenoma tissue samples compared to normal colonic mucosa.Furthermore, elevated CCAT1 levels could be detected in lymph node and distant liver metastases, as well as in peripheral blood mononuclear cells (PBMCs) of CRC patients[96]. Alaiyanet al[23]have confirmed the overexpression of CCAT1 in precancerous conditions and through all CRC stages using RT-qPCR andin situhybridization (ISH). These data suggest its essential role in both early carcinogenesis and metastatic processes, moreover,in vitrostudies revealed that the c-Myc oncogene could facilitate the transcription of CCAT1 by binding to its promoter[97]. CRNDE also becomes activated already in the initial steps of tumor development as its elevated expression was observed in > 90% of neoplastic colon tissue including adenoma and adenocarcinoma samples using both microarray and RT-PCR technology[24]. Liuet al[103]found significant upregulation of CRNDE-h splice variant both in adenoma and CRC tissues compared to control groups containing normal adjacent, inflammatory bowel disease and hyperplastic polyp samples. Moreover, within the CRC group,increased expression of CRNDE-h showed significant correlation with tumor size,lymph node, and distant metastasis. It was observed inin vitrostudies, that lncRNA CRNDE can promote CRC development and progression through epigenetic silencing of dual-specificity phosphatase 5 (DUSP5) and cyclin-dependent kinase inhibitor 1A(CDKN1A)[104]orviaactivating Ras/MAPK[105]and WNT/β-catenin[106,107]signaling pathways. Furthermore, it can contribute to chemoresistance by sponging microRNAs(miR-136[108], miR-181a-5p[107]) in CRC.
Some colorectal cancer associated lncRNAs [CRCALs: CRCAL-1 (AC021218.2),CRCAL-2 (LINC00858), CRCAL-3 (RP11-138J23.1) and CRCAL-4 (RP11-435O5.2)]were identified as overexpressed and novel CRC biomarkers using RNA-sequencing techniques[100]. These lncRNAs may “be involved in the very early steps of the neoplastic process” as the expression levels of all four CRCALs were found to be elevated in colorectal adenoma samples, as well. RNA interference-mediated knockdown experiments and gene ontology analysis of The Cancer Genome Atlas(TCGA) dataset suggest the involvement of CRCAL-3 and CRCAL-4 in cell cycle regulation[100].
Several studies have also indicated the tumor-promoting role of UCA1 lncRNA in CRC[101,102,109]. Intensive UCA1 expression was found to be correlated with larger tumor size, depth of invasion, and a less differentiated histology[110]. Moreover, elevatedUCA1 levels could be detected in precancerous adenomas which increase in CRC[102].
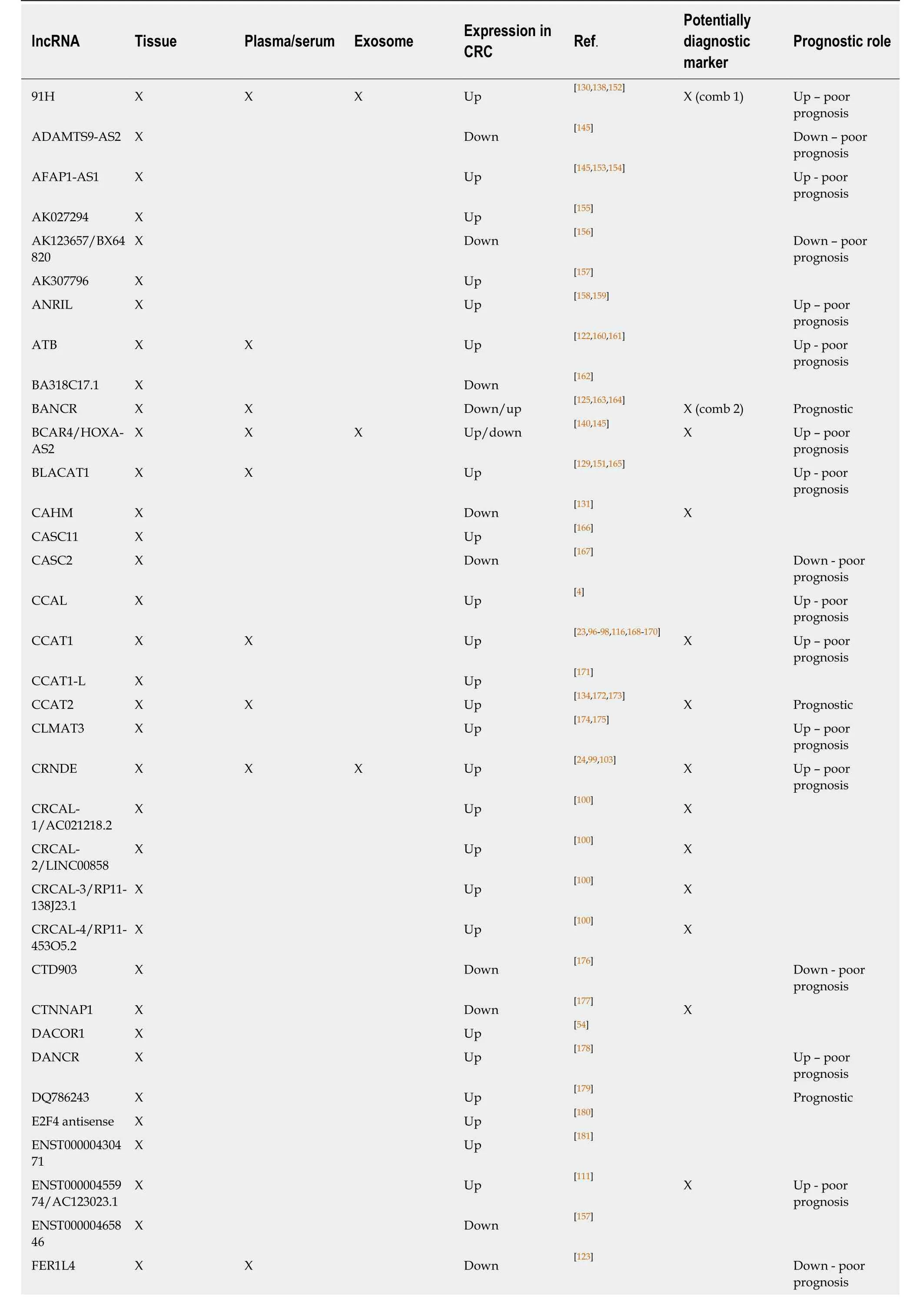
Table 1 Tissue and circulating long non-coding RNAs with altered expression in colorectal cancer
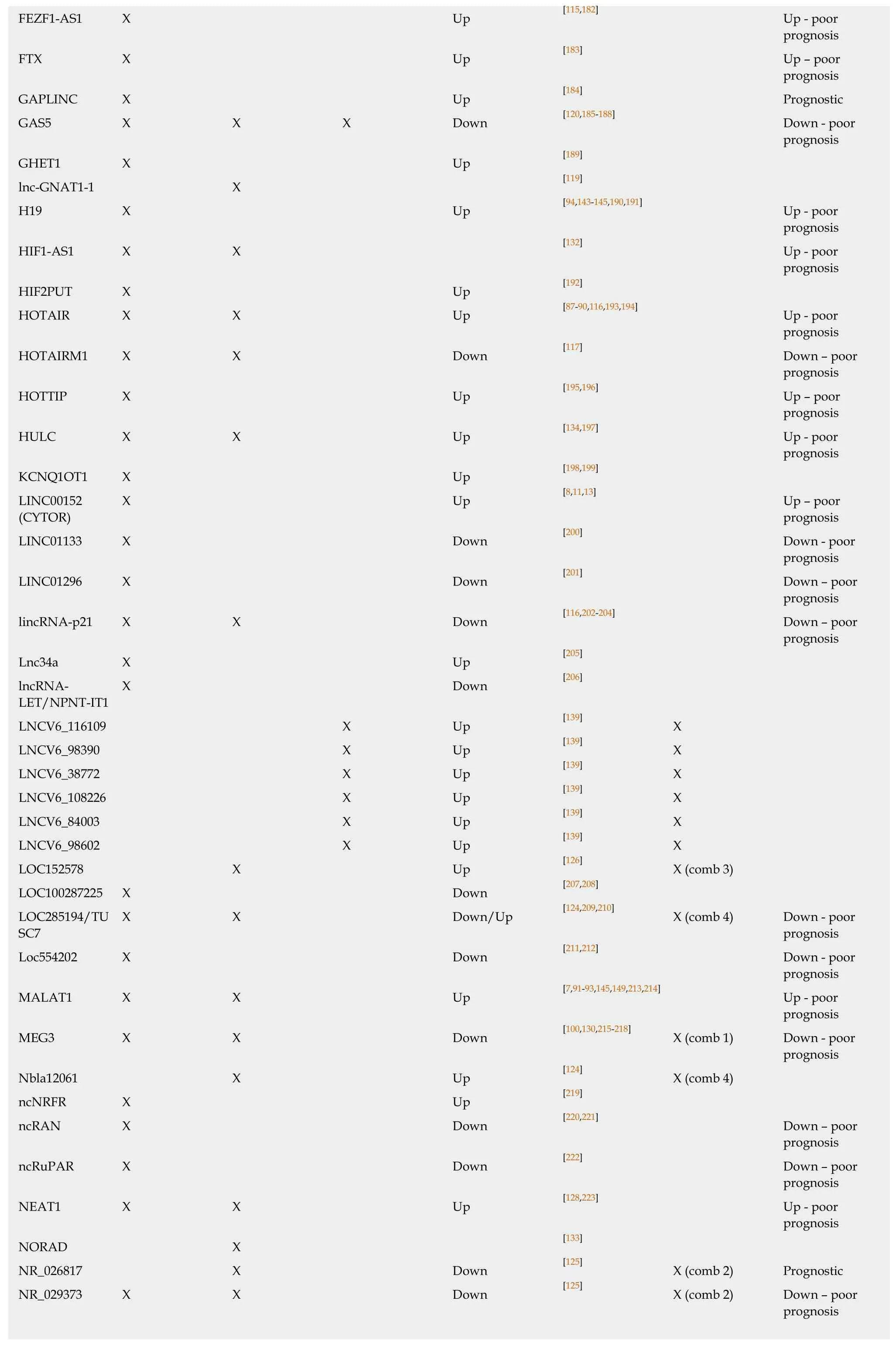
FEZF1-AS1 X Up [115,182] Up - poor prognosis FTX X Up [183] Up - poor prognosis GAPLINC X Up [184] Prognostic GAS5 X X X Down [120,185-188] Down - poor prognosis GHET1 X Up [189]lnc-GNAT1-1 X [119]H19 X Up [94,143-145,190,191] Up - poor prognosis HIF1-AS1 X X [132] Up - poor prognosis HIF2PUT X Up [192]HOTAIR X X Up [87-90,116,193,194] Up - poor prognosis HOTAIRM1 X X Down [117] Down - poor prognosis HOTTIP X Up [195,196] Up - poor prognosis HULC X X Up [134,197] Up - poor prognosis KCNQ1OT1 X Up [198,199]LINC00152X Up [8,11,13] Up - poor(CYTOR)prognosis LINC01133 X Down [200] Down - poor prognosis LINC01296 X Down [201] Down - poor prognosis lincRNA-p21 X X Down [116,202-204] Down - poor prognosis Lnc34a X Up [205]lncRNA-X Down [206]LET/NPNT-IT1 LNCV6_116109 X Up [139] X LNCV6_98390 X Up [139] X LNCV6_38772 X Up [139] X LNCV6_108226 X Up [139] X LNCV6_84003 X Up [139] X LNCV6_98602 X Up [139] X LOC152578 X Up [126] X (comb 3)LOC100287225 X Down [207,208]LOC285194/TUX X Down/Up [124,209,210] X (comb 4) Down - poor SC7prognosis Loc554202 X Down [211,212] Down - poor prognosis MALAT1 X X Up [7,91-93,145,149,213,214] Up - poor prognosis MEG3 X X Down [100,130,215-218] X (comb 1) Down - poor prognosis Nbla12061 X Up [124] X (comb 4)ncNRFR X Up [219]ncRAN X Down [220,221] Down - poor prognosis ncRuPAR X Down [222] Down - poor prognosis NEAT1 X X Up [128,223] Up - poor prognosis NORAD X [133]NR_026817 X Down [125] X (comb 2) Prognostic NR_029373 X X Down [125] X (comb 2) Down - poor prognosis
The altered expression of long non-coding RNAs in colorectal cancer tissue, plasma/serum or exosomes and the potential diagnostic value are marked with X, respectively. Combined marker sets are also represented (comb 1, comb 2, comb 3 and comb 4).
In a recent publication, Laoet al. have described the gradual elevation of expression of a novel lncRNA, AC123023.1-201 (ENST0000455974) along the colonic normaladenoma-dysplasia-carcinoma-metastasis sequence[111]. High levels of this lncRNA were found to be significantly associated with poor survival of DNA mismatch repair proficient (pMMR) CRC patients.In vitrostudies suggest that AC123023.1-201 might exert an oncogenic role in the pathomechanism of pMMR CRCviapromoting JAG2-mediated Notch signaling[111].
LNCRNA MARKERS IN PLASMA/SERUM OF COLORECTAL TUMOR PATIENTS AND THEIR MALIGNANCY-RELATED CELL FUNCTIONS
LncRNA molecules can cross the cell membrane, and hence can be found in different body fluids, such as blood, plasma/serum or urine[112]. They can be derived from apoptotic and necrotic cells, or from living cells by an active manner. These molecules occur in association with RNA-binding proteins or lipoprotein complexes, however,extracellular vesicles are reported to be the primary source of plasma lncRNAs[113].These forms contribute to the relative resistance to degradation by RNase enzymes that make circulating lncRNAs promising markers for the prognosis, diagnosis, or screening of various diseases, including CRC[114]. The altered expression levels of several lncRNAs were reported in tumor tissues of CRC patients, and recently,additional articles have been published describing their presence in plasma or serum samples[115]. CCAT1 and HOTAIR are among the first markers reported to have significantly elevated expression in the plasma of CRC patients compared to healthy controls[116]. It was also observed that after surgical treatment of CRC patients, the serum levels of these lncRNAs decreased in comparison with pre-operative samples.HOTAIR expression was also reported in peripheral blood mononuclear cells (PBMC)of CRC blood donors as compared with controls; of note, patients with right-sided CRC had lower levels of HOTAIR lncRNA than those with left-sided cancers[90]. In contrast, HOX antisense intergenic RNA myeloid 1 (HOTAIRM1) showed reduced expression in tumor tissue, and low levels were reported in plasma of CRC patients compared to healthy controls using nested TaqMan RTPCR method[117]. It has been assumed that this lncRNA can inhibit intense cell division and therefore, it may function as a tumor suppressor. The expression of lncRNA SLC25A25-AS1 was also significantly decreased in both tumor tissue and serum samples, and based onin vitromeasurements, it was observed that downregulation of SLC25A25-AS1 has an impact on chemoresistance and induces the epithelial-mesenchymal transition (EMT)process[118]. Low levels of lnc-GNAT1-1 were detected in the plasma of CRC patients,and with advanced TNM stages, the level of this lncRNA decreased in the peripheral blood[119]. LncRNA growth arrest specific transcript 5 (GAS5) had diminished expression in serum samples of 109 CRC patients compared with 99 healthy controls[120]. Further experiments highlighted that low level of GAS5 was correlated with advanced TNM stages and larger tumor size. LncRNAs that can enhance cell proliferation were also described in some reports. For instance, lncRNA SPRY4-IT1 was found to be significantly upregulated in CRC tissue and serum samples, and its increased expression was associated with late TNM stages. It influences proliferation,migration, and invasion of CRC cells, and has an effect on the expression of EMTrelated genes[121]. Long non-coding RNA-activated by TGF-β (lncRNA-ATB) has been analysed in 50 preoperative and postoperative plasma samples of cancer patients and in 50 healthy volunteers, and its overexpression was reported in 70% (35/50) of CRC cases one month after surgery[122]. Moreover, lncRNA-ATB levels were found to be significantly higher in postoperative plasma in comparison with preoperative samples, suggesting that lncRNA might be released by other mechanisms than by the primary tumor. This research group described another lncRNA, fer-1-like protein 4(FER1L4) that showed decreased expression level in postoperative blood samples compared with the matched preoperative ones in contrast to the above-mentioned lncRNA-ATB[123]. Wanget al. compiled a panel of lncRNA containing 3 RNAs(LOC285194, RP11-462C24.1, and Nbla12061) that were upregulated in 61 CRC serum samples compared to healthy controls (n= 60)[124]. Another study selected four lncRNAs (BANCR, NR_026817, NR_029373, and NR_034119) for further experiments after high-throughput microarray analysis, and concluded that this panel was dysregulated in tissue and serum samples of colon carcinoma patients[125]. Shiet al[126]also performed microarray analysis on the circulating plasma lncRNA fraction using Human LncRNA Array v3.0, and 8 transcripts were further examined with RT-qPCR technique. From these candidates, expression of three (XLOC_006844, LOC152578,and XLOC_000303) lncRNAs were found to be significantly higher in CRC plasma samples (n= 220) compared to cancer-free controls (n= 180). Another lncRNA,nuclear-enriched abundant transcript 1 (NEAT1) was identified based on microarray results as the most significantly upregulated gene in whole blood samples of CRC patients[127]. Two variants of this lncRNA, NEAT1_v1 and NEAT1_v2 were studied separately, and high levels of both two transcripts were observed[128]. Moreover, Wuet al[129]showed that knockdown of NEAT1_v1 caused inhibition of cell invasion and proliferationin vitro, while in case of NEAT1_v2, the knockdown of the transcript could induce cell growth. Similarly to the previous studies, lncRNA bladder cancer associated transcript 1 (BLACAT1) was also found to be overexpressed using microarray analysis and the increased expression was confirmed using RT-PCR in CRC serum samples. Liuet al. selected 3 lncRNAs, H19 antisense (91H), plasmocytoma variant translocation 1 (PVT-1) and maternally expressed gene 3 (MEG3)and reported increased levels in plasma of CRC patients compared to non-cancerous controls[130]. Our knowledge on the regulation of lncRNA gene expression is incomplete; however, a study by Pedersenet al[131]demonstrated reduced level of lncRNA CAHM in CRC patients coupled with elevated methylation of CAHM gene,which was detectable also in plasma samples.
Additional circulating lncRNAs have been described as potential biomarkers for CRC detection (e.g., HIF1A-AS1, NORAD, CCAT2 or HULC), and more are expected to be identified in the near future[132-134]. The most promising lncRNAs to date are summarised in Table 1.
APPEARANCE OF LONG NON-CODING RNAS IN EXOSOMES
Exosomes are a subgroup of extracellular vesicles (EVs) that can be found in different body fluids, including blood, serum/plasma, urine or saliva. The particles range from 30 to 100 nm in diameter, and around 2 × 1015exosomes have been identified in the blood of healthy people; however, in case of cancer, the exosome numbers can increase, and reaching 4 × 1015[135,136]. Recent studies highlighted that exosomes secreted by tumor cells contain DNAs, proteins, lipids, different small molecules and RNAs including lncRNAs, and these molecules may also be taken by target cells. Therefore,the contents of exosomes can influence the biological functions of the recipient cells and play an important part in long distance cell-cell communication[137].
Several differentially expressed lncRNAs in exosomes were reported in plasma/serum samples of CRC patients. According to Liuet al[103], colorectal neoplasia differentially expressed-h (CRNDE-h) showed elevated expression in isolated exosomes of 148 CRC patients compared to benign colorectal disease patients and healthy controls. Moreover, it was observed that a high exosomal level of this lncRNA correlated with both lymph node and distant metastasis and was related to low overall survival rates. Expression of exosomal lncRNA 91H also increased in CRC serum samples, which occurs at a higher level in the vesicles, than in exosome-free sera[138]. It has been also reported that the elevated expression was decreased after surgery. Based on real-time PCR results, Barbagalloet al[101]demonstrated that UCA1 in serum exosomes of cancerous patients was downregulated, while taurine upregulated 1 (TUG1) was overexpressed. Another study constructed a six-member(LNCV6_116109, LNCV6_98390, LNCV6_38772, LNCV_108266, LNCV6_84003 and LNCV6_98602) panel of plasma exosomal lncRNAs based on microarray analysis that indicated overexpression in CRC patients compared to healthy individuals[139]. The increased level was already observed in the early stages of CRC suggesting that these lncRNAs are potential markers for early detection of cancer. Donget al[140]showed that two mRNAs (KRTAP5-4 and MAGEA3) and one lncRNA (BCAR4) extracted from sera exosomes are present at a lower level in colorectal adenoma and carcinoma patients compared to healthy individuals, and the combination of these RNAs could be used as CRC biomarkers. Interestingly, according to Liet al[120]lncRNA GAS5 was found to be downregulated in CRC sera samples and acts as a tumor suppressor in cancer development, however, another study revealed that this lncRNA was upregulated in tissues, plasma and exosomes of CRC patients and its expression was related to TNM stage, Dukes stage, lymph node metastasis, local recurrence rate and distant metastasis rate[141].
Analysis of lncRNAs in exosomes is ongoing, and because altered levels of lncRNAs can serve as a potential markers for CRC detection, clarification of their function in cancer development is also a crucial step. The exosomal lncRNAs with altered expression in CRC are listed in Table 1.
CLINICAL RELEVANCE OF ALTERED LONG NON-CODING RNA EXPRESSION PATTERNS IN CRC
Biomarkers - as objectively measurable molecules suitable for monitoring physiological and pathological processes and the effect of treatments - have a crucial role in the clinical workup of tumors, enhancing the early diagnosis, classification of tumors, monitoring therapy response, and supporting the evolvement of personalized therapies, as well[21]. LncRNAs can serve as diagnostic, prognostic and predictive biomarkers in malignant diseases including CRC[22]. Principally, lncRNAs with altered levels in different stages of tumorigenesis and progression have a great potential to become early diagnostic and/or prognostic biomarkers. Besides the remarkable expression difference associated with disease stages, the important aspect of their presence and stability in the circulatory system are opening a new path for noninvasive diagnostic applications[21,142]. CCAT1 can serve as a promising marker for early CRC recognition due to its high expression in malignant and benign colorectal tumors compared to normal controls[23,96], and its detection both in PBMC and plasma samples, as well[96,116]. Increased plasma CCAT1 could predict the presence of CRC with 75.7% sensitivity and 85.3% specificity[116]. Almost all splice variants of CRNDE lncRNA, (except for CRNDE-d), and particularly CRDNME-b and CRNDE-h, were found to be intensively (approximately 5- to 100-fold) upregulated in both benign and malignant neoplastic colorectal tissue[24]. On the basis of CRNDE-h expression levels,CRC and normal tissue samples could be discriminated with 85% sensitivity and 96%specificity, which was also proven to be a highly sensitive and specific marker in adenomavsnormal tissue comparison (sensitivity: 95%, specificity: 96%)[24]. Based on CRNDE-h levels in tissue, CRC could be differentiated from adenoma and healthy tissues with 70.4% sensitivity and 70.8% specificity[99]. Its strong diagnostic potential was also supported by the circulating CRNDE-h RT-qPCR results at a reported 87%sensitivity and 93% specificity between CRCvshealthy controls[24]. Moreover, the analysis of exosomal CRC-related CRNDE-h of serum also allowed separation of CRC samples from benign and healthy controls (AUC = 0.892, sensitivity: 70.3%,specificity: 94.4%)[103]. The newly identified upregulated CRCAL1-4 lncRNAs might be suitable for early recognition of colorectal neoplasias, however, only marginal significance could be observed between adenoma and CRC[100]. Potential utilization in CRC screening and diagnostics of several other differentially expressed lncRNAs including BLACAT1[129], CCAT2[134], HULC[134], NEAT1[128], UCA1[101,109]and HOTAIRM1[117]has also emerged in RT-qPCR studies analyzing circulating lncRNAs resulting in various specificity (43%-96%) and sensitivity (55%-100%) values. In addition to the altered expression levels, the DNA methylation changes of lncRNAs can hold a discriminative ability, as the amount of methylated CAHM DNA molecules in the circulatory system depends on the CRC stages; hence it can serve as a promising marker for CRC screening[131].
In addition to single lncRNA marker candidates, lncRNA marker combinations and multi-marker lncRNA panels have also been identified as a potential diagnostic approach. By testing the CRC diagnostic efficacy of circulating HOTAIR and CCAT1,the combined measurement of their plasma/serum levels resulted in higher sensitivity and specificity values (84.3% and 80.2%, respectively) than the abovementioned markers alone[116]. This marker combination could provide an effective CRC diagnosis performance, moreover, it could detect CRC efficiently already at an early stage (85%). Analysis of Barbagalloet al[101]revealed that diagnostic accuracy of serum exosome UCA1 levels for CRC (sensitivity: 100%, specificity: 43%) could be enhanced by applying it in combination with TUG1 lncRNA (sensitivity: 93%,specificity: 64%) or with circHIPK3 circular non-coding RNA (sensitivity: 100%,specificity: 70%). A promising lncRNA panel containing three lncRNAs (LOC152578,XLOC_000303, and XLOC_0006844) upregulated in CRC was identified and validated on a large independent plasma sample cohort (220 CRCs, 180 controls) (positive predictive value: 0.80, negative predictive value: 0.84, AUC = 0.975)[126]. The doubleblind test on another 100 plasma samples (50 CRC, 50 cancer-free controls) also confirmed that the above-mentioned biomarker set is suitable for indicating the occurrence of CRC with 85% accuracy[126]. CRC and healthy normal cases could be distinguished based on the increased serum levels of LOC285194, RP11-46C24.1, and Nbla12061 lncRNAs (AUC = 0.793, sensitivity: 68.33%, specificity: 86.89%)[124]. The predictive value of this lncRNA signature was significantly higher than of the conventional clinical serum protein markers (CEA, CA199, CA125, and CA724) (AUC values were 0.633, 0.567, 0.517 and 0.592, respectively)[124]. Microarray analysis of CRC-NAT tissue sample pairs revealed a four-lncRNA panel (upregulated BANCR and downregulated NR_026817, NR_029373, NR_034119) which had consistently altered pattern both in CRC tissue and serum samples compared to normal controls[125]. The high AUC, specificity and sensitivity values for both the training and validation sample sets support the reliable diagnostic ability of this biomarker set(AUC: 0.891 and 0.881; specificity: 80% and 75.83%; sensitivity: 81.67% and 89.17%)which even exceeded the diagnostic power of CEA[125]. A pilot study of Liuet al.revealed a new promising diagnostic plasma ncRNA biomarker set (H91, PVT-1,MEG3) for early-stage CRCs as the panel could differentiate CRC samples from controls with 82.76% sensitivity and 78.57% specificity[130].
According to the Lnc2Cancer 2.0 database (www.bio-bigdata.com/lnc2cancer), the most frequently described lncRNAs with prognostic value in CRC are H19[95,143-145],CRNDE[99,103,105,107,146], HOTAIR[89,90,147,148]and MALAT1[92,145,149](Supplemental Table 1).In silicolncRNA expression analysis of CRC data from The Cancer Genome Atlas(TCGA) database (n= 534) showed that H19 was the lncRNA mostly associated with the overall survival (OS) of CRC patients (P= 0.0005), independently from tumor stages[143]. Elevated H19 levels were found to be correlated with tumor differentiation and advanced TNM stage[144], and its expression could be considered as an independent predictor for OS and disease-free survival (DFS). Other studies also confirmed that overexpression of H19 lncRNA could predict the unfavorable prognosis in CRC[145]. CRNDE-h can serve as a promising early diagnostic biomarker for CRC, and it also has a prognostic capability due to its high tissue and serum exosome levels significantly correlated with tumor size, lymph node, and distant metastasis[99,103]. In addition, increased exosomal CRNDE-h levels were proven to be a negative predictor of OS of CRC patients [34.6% (high CRNDE-h)vs68.2% (low CRNDE-h),P< 0.001)][103]. Similar associations with CRC stages were reported for CRNDE-p, another overexpressed transcript variant of CRNDE[146]. HOTAIR lncRNA was also observed to be a negative prognostic factor in CRC, as its upregulated expression in primary tumor tissue, even more in blood of CRC patients were found to be associated with higher mortality [Cox's proportional hazard, hazard ratio (HR)(tissue) = 4.4, HR (blood) = 5.9][90]. Significant differences in clinicopathological parameters such as less differentiated histology, greater tumor depth, and liver metastasis were observed in CRC cases with high HOTAIR expression (n= 20)compared CRCs with low HOTAIR levels (n= 80) (P< 0.05)[89]. Results of several other studies verified the correlation of higher HOTAIR levels with poorer OS[89,148]. With the RT-qPCR analysis of tissue samples from 146 stage II/III CRC patients, it was observed that patients with more intense MALAT1 lncRNA expression had a significantly worse prognosis with a HR of 2.863 for DFS and 3.968 for OS[92].Moreover, high MALAT1 levels were found to be associated with decreased patient survival and poor response to oxaliplatin-based chemotherapy in advanced CRC patients suggesting its utility as a prognostic marker and therapeutic target in CRC[149].
In addition to CRNDE[103,146]and HOTAIR[90,116], among the 31 potentially prognostic lncRNAs published in at least two independent studies, CCAT2[150], GAS5[141],BLACAT1[129], CCAT1[96,116], NEAT1[128], 91H[138]and BANCR[125]lncRNAs were also detectable in the circulation suggesting their application as minimally invasive markers for CRC prognosis (Table 1 and Supplemental Table 1). As reported by Ozawaet al[150]in a study involving two independent cohorts, the evaluation of CCAT2 expression in combination with CCAT1 may be a powerful tool for predicting tumor recurrence and prognosis in CRC patients. According to the expression analysis in tissue, plasma and exosome samples, GAS5 had a prognostic value in CRC based on its expression that was negatively correlated with TNM status, Dukes stage,and lymph node metastasis (LNM), local recurrence and distant metastasis rate, while its level was in positive relation with differentiation degree and the 3-year OS rate[141].On the other hand, elevated BLACAT1 expression could be considered as an independent unfavorable prognostic indicator for CRC, as it was observed to be associated with advanced CRC stages and shorter OS[151]. The predictive potential of lncRNA transcript variants can differ, as the OS of CRC patients with intensive NEAT1_v1 expression was worse, while high levels of the other isoform, NEAT1_v2 was correlated with better OS[128]. Determination of clinical significance of elevated exosomal H91 lncRNA expression suggested that it might be an early minimal invasive biomarker for CRC recurrence or metastasis[138]. Gonget al[132]evaluated the diagnostic and prognostic value of increased serum HIF1A-AS1 levels in 151 CRC and 160 healthy control samples by RT-PCR, and reported a high diagnostic efficacy(86.8% sensitivity and 92.5% specificity); moreover, it was described as a predictor for worse prognosis in CRC.
In addition to the diagnostic and prognostic utility of lncRNAs with altered expression, ongoing research focused on the role of lncRNAs in chemoresistance and therapy response prediction are revealing several lncRNAs which could be promising therapeutic targets in CRC. Similarly to the above-mentioned MALAT1 whose increased levels were found to be associated with poor response to oxaliplatin (OXA)-based chemotherapy[149], CRNDE can also contribute to oxaliplatin resistance in CRC[107,108]. According to a recentin vitrostudy, CRNDE facilitates the resistance against OXA or 5-fluorouracil (5FU) treatmentviamiR-181a-5p-mediated regulation of Wnt/β-catenin signaling[107]. Association between high HOTAIR expression and poor response to 5FU treatment was assessed[147]. HOTAIR can contribute to 5FU resistance through suppressing miR-218 and activating NF-κB signaling in CRC[147].HOTAIR was observed to be upregulated in drug-resistant cisplatin- or paclitaxeltreated SW620 and Colo205 CRC cells, as well[148]and could affect the chemoresistance of CRC via miR-203a-3p-mediated modulation of Wnt/β-Catenin pathway[148]. The most important tissue and circulating lncRNAs with diagnostic and prognostic potential in colorectal tumors are represented in Figure 1.
CONCLUSION
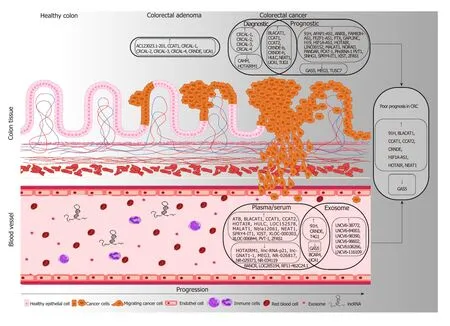
Figure 1 The most important tissue and circulating long non-coding RNA candidates with diagnostic and prognostic potential in colorectal tumors. Long non-coding RNA (lncRNAs) upregulated in adenoma or colorectal cancer (CRC) samples compared to normal controls are marked with ↑, while the downregulated lncRNAs are depicted with ↓. Potential prognostic markers detectable both in tissue and blood specimens are highlighted in the right, where ↑ refers to lncRNAs whose higher levels were found to be associated with poor prognosis (CRNDE, HOTAIR, CCAT2, BLACAT1, CCAT1, NEAT1, 91H, HIF1A-AS1), while the low expression of lncRNA marked with ↓(GAS5) can be a predictor of worse disease outcome in CRC patients. In case of the lncRNAs written without frame (BANCR,BCAR4, LOC285194, RP11-462C24.1, UCA1), diverse, sometimes controversial expression data are available in the scientific literature.
The increasing number of genome-wide expression analysis studies have led to the identification of a number of long non-coding RNAs with altered expression patterns in cancers including CRC. LncRNAs are proven to contribute to each step of the colorectal carcinogenesis and tumor progression by influencing the key cancer-related signal transduction pathways such as WNT/β-catenin, PI3K/Akt, EGFR, NOTCH,mTOR and TP53 signaling. Dysregulated lncRNAs can appear in the pre-malignant adenoma stage of CRC and the expression alterations of a relatively large number of lncRNAs were found to be associated with clinicopathological parameters indicating CRC progression. Furthermore, lncRNAs are stable and detectable in body fluids facilitating their utilization as early detection and prognostic biomarkers. In order to open the door for implementation of minimally invasive lncRNA-based tests in the clinical practice, certain relevant technical aspects should be considered: (1)Standardization of the pre-processing and sample preparation procedure including the applied blood collection tubes, sample storage conditions and time, optimized lncRNA isolation protocols from liquid biopsy samples; (2) Selection of appropriate quantification, quality checking and sensitive techniques allowing the precise detection of cancer-related alterations; and (3) Application of proper universal endogenous controls for increasing the reliability and the accuracy of RT-qPCR measurements. For the development of adequately sensitive and CRC-specific,clinically applicable diagnostic and prognostic tests based on lncRNA markers/marker panels, validation studies with large sample cohorts are essential.On the other hand, as recent studies shed light on the potential role of lncRNAs as novel therapeutic targets, the specific lncRNA expression alterations in liquid biopsy samples may contribute to the improved early recognition, prognosis prediction and therapy monitoring in CRC. Moreover, lncRNAs as druggable targets might represent the basis of novel therapeutic methods in the fight against cancer.
ACKNOWLEDGEMENTS
We thank Ramani Gopal PhD and Theo deVos PhD for their careful language assistance.
杂志排行

World Journal of Gastroenterology的其它文章
- Autoantibodies: Potential clinical applications in early detection of esophageal squamous cell carcinoma and esophagogastric junction adenocarcinoma
- Role of endoscopic ultrasound in the screening and follow-up of high-risk individuals for familial pancreatic cancer
- Hepatic senescence, the good and the bad
- Optimizing proton pump inhibitors in Helicobacter pylori treatment:Old and new tricks to improve effectiveness
- Regulatory effect of a Chinese herbal medicine formula on nonalcoholic fatty liver disease
- Allyl isothiocyanate ameliorates lipid accumulation and inflammation in nonalcoholic fatty liver disease via the Sirt1/AMPK and NF-κB signaling pathways